
2974
Views & Citations1974
Likes & Shares
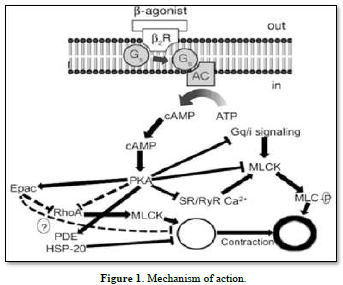
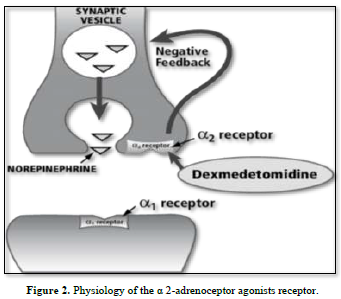
Mechanism of action of b 2-adrenoceptor agonists. AC adenylyl cyclase, b 2 R b 2 adrenoceptor, cAMP cyclic adenosine monophosphate, Epac exchange protein directly activated by cAMP, G s stimulatory G-protein, HSP-20 heat shock-related protein 20, MLCK myosin light chain kinase, MLC-P myosin light chain phosphatase, PDE phosphodiesterase, PKA protein kinase A, SR/RyR Ca 2? sarcoplasmic reticular ryanodine Ca. It is well known that these receptors can be utilized therapeutically to treat functional abnormalities of the lower urinary tract since stimulation of these receptors increases intraurethral pressure while inhibition decreases it [13]. Certain medications have been proposed to have specificity for Studying the a-adrenoceptor subtypes in this area is important since urethral a-adrenoceptors can be acquired [14]. Therefore, the current study's objectives were to use a radioligand binding technique to quantify the populations of al- and a2-adrenoceptors in the female rabbit bladder base and urethra and to examine the functional role of al-and/or 12-adrenoceptors located postjunctional in urethral contractions (Figures 1 & 2).
TECHNIQUES
Analysis
The bladder and urethra of female Danish Land race rabbits, weighing roughly 3 kg, were removed in one piece after the rabbits were slaughtered. The urethra was carefully dissected free from the vaginal wall down to the urethrovaginal junction and removed, beginning at the base of the bladder. Studies on radioligand binding preparation of membranes. The mucosa, periurethral fat, and connective tissue were removed by cutting open the rabbit bladder base and urethra. The leftover tissue was preserved for up to a month by freezing it in a 0.9% w/v NaCl solution. In order to perform binding tests, pooled tissues were finely chopped and at ambient temperature. After passing the homogenate through a 0.5 mm nylon mesh filter, it was centrifuged for 20 min at 4°C at 39,000 x g. After being suspended in new buffer, the pellet was recently centrifuged. The final pellet was then reconstituted in new buffer and filtered through a 0.118 mm nylon mesh; the binding experiments were conducted using the filtrate. Assay for binding Prazosin, rauwolscine, and tritium-labeled dihydro-ergocryptine ([3H]-DHE) were employed 3H]-DHE test. The [3H]-DHE binding was carried out according to the earlier instructions. In summary, a total volume of 0.5 ml was incubated for 60 min at 25°C with membranes, [3H]-DHE, drug solutions, or their solvents, in triplicate for saturation experiments and in duplicate for inhibitory tests. Diluting was used to end the incubation. homogenized in an ice-cold buffer containing 50 mM Tris-HCl (pH 7.5) for 4 x 8 s (setting 7) using a Polytron PT 10/35 homogenizer (Kinematica). the samples in 1.5 milliliters of ice-cold buffer, and then quickly filter them under low pressure using Whatman GF/C glass fiber filters that had been previously soaked in a solution containing 1 mg per milliliter of bovine serum albumin. Four quick washes of the filters were performed using 5 ml pieces of ice-cold buffer. fH]-prazosin assay Membranes were incubated in triplicate in a total volume of 0.5 ml at 25°C for 15 min with [3H]-prazosin, redistilled water, or phentolamine (10-I M). Three 5 ml washes were conducted once the reaction was stopped as instructed for the [3H]-DHE test. Assay for [3H]-rauwolscine Membranes, redistilled water, [3H]-rauwolscine, and either phentolamine (10-5M) were incubated in triplicate in a 0.25 ml total volume at 25°C for 20 min. The reaction was halted in accordance with the [3H]-DHE protocol. test, with the exception that the filters were pre-soaked in buffer devoid of bovine serum albumin and the samples were diluted with one milliliter of ice-cold buffer. Using a liquid scintillation spectrometer (LKB RackBeta 1215), the tritium content was measured after the filters were put in scintillation vials. Using the external standard channels ratio approach, it was found that the counting efficiency was approximately 50%. Early association studies using the 3Hligands revealed that binding was persistent for at least an extra fifteen minutes after each ligand's incubation time had achieved steady state. Membranes were added to begin each reaction, which was then allowed to run for 60, 15, and 20 min for [3H]-DHE, [3H]-prazosin, and [3H]-rauwolscine, respectively. Following that, the filtration process was initiated and done in five to fifteen minutes. Each tritiated substance's specific binding was defined as that which was greater than the binding in blanks containing 10-5 M phentolamine. An aliquot of 20 yards was extracted from many tubes and its tritium activity quantified in order to calculate the quantity of radioactive ligand in the test. By measuring the difference between added and bound 3H-ligand, it was possible to estimate the concentration of free radioligand in the saturation tests. The protein content was measured using the Lowry [15] method, which used 50 mM Tris-HCl buffer as a blank and bovine serum albumin as a reference. Dixon's gap test was used to look for outliers. For computations, the mean values from duplicate or triplicate determinations were utilized. The lines' linear least square fit as appropriate, was applied to the different plots. Ki values were calculated using Cheng & Prusoff's [16] approach. Study functions the internal urethral opening and the trigonal area were located once the bladder was opened. The middle and upper sections of the urethra were cut into two circular transverse sections, each measuring 2-4 mm in length. After each ring-formed preparation was placed in an organ bath that was kept at a constant temperature of 37°C using Krebs solution (see below for composition), it was bubbled with a mixture of 95% CO2 and 5% 2% alumina to bring the pH down to about 7.4. The preparation was positioned in the urethral ring lumen between two L-shaped steel hooks. For isometric registration, one hook was attached to a force transducer (Statham FT 03). tension. The other hook was fastened to a moveable component that let the tension be changed. After being stretched repeatedly, the urethral rings were allowed to relax until a steady tension of about 6 mN was reached. The preparations were subjected to 6.2 x 10-5 M noradrenaline (NA), supplied non-cumulatively, after a 60-min equilibration period. It was discovered in separate studies that this exposure produced 90±2% of the highest response achieved when NA was added cumulatively. Using a cumulative approach, concentration-response curves for phenylephrine (PE), clonidine, and NA were produced. A single concentration of agonist induced a submaximal contractile response, which was used to investigate the effects of a.-adrenoceptor blockers. To ensure repeatability, agonist doses below the submaximal range were employed. The medication that blocks was added. Two consecutive, repeatable responses with less than 10% fluctuation were obtained; the antagonist's contact time was between 10 and 20 min. Once a maximum response was achieved, the drugs were removed. Worksheets The mean ± s.e. mean is provided as the results. The curves were used to graphically determine the IC50 and EC50 values. In functional investigations, n is the number of urethral rings evaluated; in binding studies, n is the number of experiments. Remedies and medications Studies that bind [3H]-dihydro-a-ergocryptine was diluted in an aqueous solution with 40% ethanol, resulting in an assay tube final concentration of roughly 1.6% ethanol. Diluted [3H]-rauwolscine was added to 1 mM HCI containing 20% ethanol, yielding assay tubes containing around 1.6% ethanol. 1 mM HCI was used to dilute [3H]-prazosin. Because of the Due of prazosin's difficulty dissolving, two stock solutions comprising 2.5 x 10-3 and 2.5 x 10-4M prazosin were created by adding a little amount of methanol and gently heating 1 mM HCI. Prazosin has a propensity to precipitate at room temperature in the highest concentration, thus it was briefly heated before being introduced to the assay tubes. It was not noticed at the lower concentration. The two highest concentrations of prazosin utilized in the experiment were prepared using the high concentration stock solution; the remaining concentrations were prepared using the low concentration stock solution. Redistilled water was used to make all of the dilutions from these stock solutions. As previously mentioned, the impact of NA on [3H]-DHE binding was ascertained. Every other medication was dissolved in redistilled water and then serially diluted. Study functions the following components of the Krebs solution (mM) were present in the functional studies: NaCl119, KCl4.6, CaCl21.5, MgCl21.2, NaHCO3 15, NaH2PO4 1.2, and glucose 11. While the other medications were dissolved in a 0.9% w/v NaCl solution (saline), prazosin was dissolved in ethanol. Saline was used to create further dilutions of the medications, including prazosin. The pharmaceuticals and radiochemical [3H]-dihydro-a-ergocryptine (21.9-30.5 Ci mmol-1), [3H]-rauwolscine (84.4 Ci mmol-1) and [3H]-prazosin (20.2Cimmol-1) were obtained from Amersham and New England Nuclear, respectively. The following medications were also used: clonidine HCl (Boehringer Ingelheim), (-)-noradrenaline HCl (Aldrich), (±)-noradrenaline, (-)-phenylephrine (Sigma), rauwolscine HCI (Roth), prazosin HCI (Pfizer), and phentolamine methane sulphonate (Ciba-Geigy). The compounds utilized were analytically graded. Every solution was made using redistilled water.
Studies with bindable results
Biphasic displacement curves were produced when membranes derived from the female rabbit bladder base and urethra were treated with the selective ac-adrenoceptor antagonist prazosin and the selective c2-adrenoceptor antagonist rauwolscine to prevent specific [3H]-DHE binding. A plateau was obtained in the prazosin displacement curve between 10-8 and 10-6 M concentrations. The level of the plateau was calculated to be 22 ± 1% inhibition based on the mean values of the inhibition obtained at these concentrations. In a similar vein, the rauwolscine inhibition curve plateaued at 81 ± 2% inhibition between doses of 3 x 10-7 and 10-M. The following potency ranking of a- (-)-phenylephrine (log Ki= -5.49 ± 0.09; n = 5) > - 6.36 +0.20 (Figure 2). When any of these agonists or antagonists were administered at high quantities with 10–5 M phentolamine, there was no further inhibition of [3H]-DHE binding observed when compared to separate applications of 10–5 M phentolamine, indicating that these drugs compete for the same binding sites. A 25 + 4% (n = 7) suppression of binding was observed when the displacement of specific [3H]-DHE binding was assessed in the presence of 3 X 10-8 M prazosin. At a dose of 3 x 10-7M, the equivalent inhibition for rauwolscine was 75 ± 3% (n = 7). When these two compounds were combined and incubated at the quantities previously specified (using the identical membrane preparation as rauwolscine and prazosin), adrenoceptor agonists were observed in their competition for specific [3H]-DHE binding: clonidine (log Ki= -6.96+0.18; n = 5) > (-)-adrenaline (log Ki=) displacement), n = 7 had a specific binding displacement of 93 ± 3%. The number of cx-adrenoceptors and xland CX2-adrenoceptors in the rabbit bladder base and urethra was ascertained by saturation studies using [3H]-DHE, [3H]-prazosin, and [3H]-rauwolscine. Scatchard plots showed that [3H]-DHE, a non-selective a-adrenoceptor blocker, binds to 76 ± 7 fmol mg-1 protein, which is a saturable number of binding sites. A finite number of receptors were found for both [3H]-prazosin and [3H]-rauwolscine when the results of saturation tests were treated similarly (Table 1, Figure 3). The values of B, for these drugs were 19 ± 4 and 5 1 ± 6 fmol bound/mg protein, respectively. The Scatchard plots for the particular binding of [3H]-prazosin and [3H]-DHE and [3H]-rauwolscine were all linear, indicating that the ligands had an equal affinity for the specific receptors to which they were bound, indicating binding to a single population of binding sites. Agonist concentration (M) =.0 75 C -0 75 I 0 50 C 25 0.r_ C n 10 8 10 7 10 6 lO 5 Figure 2: A-adrenoceptor agonists such as clonidine (0), (-)-noradrenaline (O), and (-)-phenylephrine (-) inhibit the binding of [3H]-dihydro-a-ergocryptine ([ H]-DHE). The agonists were added to membrane suspensions at the given concentrations while they were incubated with 1 nrm H]-DHE. For each agonist, the data are displayed as a percentage of the specific binding inhibition (n = 5). Study functions the selective ol- and M2-adrenoceptor agonists, PE and clonidine, respectively, as well as the non-selective a-adrenoceptor agonist NA induced the rabbit urethra's contractile responses (Figure 4). Clonidine-induced contractions grew more slowly than NA and PE-induced contractions, and there was a slower rate of relaxation following agonist wash-out (Figure 4). The EC50 values for clonidine (n = 6) and PE (n = 6) were 2.3 x 10-7 M and 1.2 x 10-5 M, respectively. While the maximal response elicited by clonidine was lower (73 ± 9%; n = 6) than that to NA, PE produced the same maximum contractile response (105 ±9%; n = 6) as 6.2 x 10-5 M NA. The study examined the inhibitory effects of rauwolscine (1.5 x 10 -6M) and prazosin (10-6 M) on contractions induced by a single concentration of NA (7.8 x 10-6). M). Rarawolscine decreased the NA-induced contractions by 52 ± 4% (n = 7) but prazosin depressed them by 84 ± 7% (n = 6). The same quantities of prazosin and rauwolscine were also tested for their ability to suppress contractions caused by a single PE concentration (7.9 x 10-6 M). These contractile reactions to PE were shown to be reduced by prazosin (89 ± 4%; n = 6) and rauwolscine (34 ± 5%; n = 11). The equivalent values for prazosin and rauwolscine were 65 + 8% (n = 5) and 69 ± 5% (n = 5) for contractions generated by clonidine (10-6M), respectively. Nevertheless, rauwolscine eliminated the responses (n = 6) when clonidine (10-7M) at a lower concentration was used to generate contractions, although only 13±4% of the contraction occurred when prazosin was present was blocked (n = 6) (Figure 3).
Conversation
It is commonly known that when different concentrations of the highly selective adrenoceptor antagonist prazosin are used to inhibit the binding of a fixed concentration of [3H]-DHE, the drug can produce biphasic inhibition curves [5,17-20]. The two phases of the inhibition curve may be taken to represent the displacement of [3H]-DHE from the al- and c2-adrenoceptors, respectively, since it is claimed that [3H]-DHE binds to ca1-and cz2-adrenoceptors with comparable affinity [5,20]. The percentage of the subtype to which the ligand is selective, relative to the entire receptor population that the 3H-ligand occupies, will therefore be represented by the plateau of the inhibition curve. According to Hoffman [5], prazosin has such a strong ability to discriminate between the ax- and a2-adrenoceptors in the rabbit uterus that it is possible to graphically estimate the proportion of each subtype and the ligand's dissociation constant from the displacement curve alone. However, due of the partial overlap of the two components, visual estimations derived from the Yohimbine displacement curve would not yield accurate estimates. We employed rauwolscine as a selective c2-adrenoceptor antagonist in our research since it has been shown to have a greater affinity for 02-adrenoceptors than yohimbine [4,9,22]. When prazosin was applied to prevent particular [3H]-DHE binding to unrefined membrane preparations derived from the base of the rabbit bladder and nicely defined, biphasic displacement curves were acquired for the urethra. The plateau's level revealed that 22% of the cx-adrenoceptor population were c1-adrenoceptors, and as a result, 78% of the a2-adrenoceptor population. The IC50 value for the first phase (adrenoceptors) was roughly 2.1 x 10-9M, as determined visually from the mean curve, while the corresponding value for the second phase (adrenoceptors) was approximately 3.9 x 10-SM. Therefore, in these trials, prazosin seems to have an 18,500-fold higher affinity for al-than M2-adrenoceptors. A Ki value of roughly 0.9 nM is produced by applying Cheng & Prusoff's [16] equation to the IC50 value for prazosin at the a1-adrenoceptor. This value is comparable to the KD value of 0.4 nM displayed for [3H]- prazosin in. At the rauwolscine plateau Rauwolscine is not as selective for c2-adrenoceptors as prazosin is for al-adrenoceptors, as seen by the less pronounced inhibition curve compared to that of prazosin. The rauwolscine inhibition curve's plateau level indicated that 81% of all a-adrenoceptors were aC2-adrenoceptors, meaning that 19% of a-adrenoceptors were cl-adrenoceptors. This is consistent with the prazosin levels that were discovered. A visual estimate of the discriminating power by rauwolscine for the al- and M2-adrenoceptor subtypes was about 900 times. As previously calculated, the Ki value for rauwolscine at M2-adrenoceptors was 8 nM, which is the same as the KD value of 8 nm for [3H]-rauwolscine in. It makes sense to Since the inhibitory effects of prazosin (3 x 10 8M) and rauwolscine (3 x 107M) were practically additive under the assay conditions specified, it is reasonable to believe that these drugs displaced distinct subdivisions of the total oa-adrenoceptor population labeled by [3H]-DHE. Consequently, we deduce that the inhibition of the corresponding ax-adrenoceptor subtype is represented by the two phases of the prazosin and rauwolscine inhibition curves. Saturation studies with [3H]-DHE, [3H]-prazosin, and [3H1-rauwolscine provided more proof. Based on the overall number of binding sites, [3H]-prazosin makes up 27% and [3H]-rauwolscine 73% of the total number of receptors that these two medications have tagged; this total nearly matches the number of receptors reported for [3H]-DHE. These ratios of alpha- and beta-adrenoceptors match the values that were previously inferred from the displacement curves. Based on research on radioligand binding, it is challenging to pinpoint the labeled receptors' exact cellular position. Since oc-adrenoceptors are known to be present on several sites, such as blood vessels and adrenergic and cholinergic nerve terminals, it is likely that some of these sites, along with adrenoceptors on the smooth muscle, were labeled during these investigations. We thus investigated a few responses of the female rabbit urethral smooth muscle in vitro in order to investigate the postjunctional positioned a-adrenoceptors. Urethral smooth muscle contractions were elicited by NA, the xl-adrenoceptor selective agonist PE, and the 02-adrenoceptor selective agonist clonidine. It is well known that PE and clonidine are not as selective for their respective a-adrenoceptor subtype as the antagonist’s prazosin and adrenoceptor M2-adrenoceptors, despite the fact that these findings imply that both of these receptors are positioned postjunctional and are implicated in the contractile response. Thus, studies using the latter medications were conducted using rauwolscine. The results showed that prazosin nearly completely eliminated the submaximal contractions caused by PE, but had less of an impact on the contractions caused by clonidine (10-6 M); when it came to NA, prazosin's inhibitory effect fell somewhere in between the first two values. Nevertheless, the potency order was inverted when rauwolscine was employed as the antagonist. Moreover, rauwolscine eliminated the contractile effect of clonidine at a concentration of 10-7 M, which is thought to be "more selective," while prazosin only partially blocked similar contractions. Therefore, it appears that postmutational both ol- and o2-adrenoceptors are present and both mediate contractions. It is frequently challenging to draw a precise comparison between binding and functional research. Apart from distinct test parameters in binding assays, agonists are often shown to interact with [3H]-DHE labelled adrenoceptors in a manner that produces concentration-inhibition curves with shallow slopes. The oa-adrenoceptors labeled by [3H]-DHE were observed to interact with the agonists NA, PE, and clonidine in the current investigation, producing flat concentration-inhibition curves. This may be explained by the agonist's varying affinities for the various x-adrenoceptor subtypes, for example, or by the receptor's potential for a negative cooperative interaction. As an alternative, the agonists may bind to one receptor subtype's high and low affinity binding sites. Hoffman [20] reported on this for the al-adrenoceptor, while Stiles [23] raised doubts about it. An agonist's interaction with the M2-Guanine nucleotides have been demonstrated to have an impact on the adrenoceptor [20]. It's possible that a single cell may control these nucleotide concentrations and, consequently, how responsive it is to activation of oa-adrenoceptors. The observation that around 75% of the M2-adrenoceptors in our membrane homogenate may account for the shallow agonist inhibition curves we observed. Moreover, neither neuronal nor extra neuronal NA uptake blockers nor 13-adrenoceptor blockers were employed in the functional experiments, despite the fact that their presence would likely change the EC50 values.
CONCLUSION
Crude membrane preparations from the rabbit bladder base and urethra have been discovered to include a-adrenoceptors, which are composed of around 25% ml-adrenoceptors and 75% M2-adrenoceptors. Some of the adrenoceptors, it seems, are situated postjunctional on the urethral smooth muscle and that contractions can be induced by any variety.
- Langer SZ (1974) Presynaptic Regulation of Catecholamine Release. Biochem Pharm 23: 1793-1800.
- Berthelsen S, Petringer WA (1977) A Functional Basis for Classification of a-Adrenergic Receptors. Life Sci 21: 595-606.
- Wikberg JES (1979) The Pharmacological Classification of Adrenergic a1 and a2 Receptors and their Mechanisms of Action. Acta Physiol Scand Suppl: 468.
- Starke K (1981) A-Adrenoceptor Subclassification. Rev Physiol Biochem Pharm 88: 199-236.
- Hoffman BB, De Lean A, Wood CL, Shocken DD, Lefkowitz RJ (1979) Alpha-Adrenergic Receptor Subtypes: Quantitative Assessment by Ligand Binding. Life Sci 24: 1739 -1746.
- De Mey J, Vanhoutte PM (1981) Uneven Distribution of Postjunctional Alpha1- and Alpha2-Like Adrenoceptors in Canine Arterial and Venous Smooth Muscle. Circ Res 48: 875-884.
- Ruffolo Jr., RR, Waddell JE, Yaden EL (1981) Postsynaptic Alpha-Adrenergic Receptor Subtypes Differentiated by Yohimbine in Tissues from the Rat. Existence of Alpha-2 Adrenergic Receptors in Rat Aorta. J Pharm Exp Ther 217: 235-240.
- Skarby TVC, Andersson K-E, Edvinsson L (1983) Pharmacological Characterization of Postjunctional Ax-Adrenoceptors in Isolated Feline Cerebral and Peripheral Arteries. Acta Physiol Scand 117: 63-73.
- Langer SZ, Massingham R, Shepperson NB (1980) Presence of Postsynaptic a2-Adrenoreceptors of Predominantly Extrasynaptic Location in the Vascular Smooth Muscle of the Dog Hind Limb. Clin Sci 59: 225-228.
- Timmermans PBMWM, Van Zwiethn PA (1980) Vasoconstriction Mediated by Postsynaptic a2- Adrenoceptor Stimulation. Naunyn-Schmiedebergs Arch Pharm 313: 17-20.
- Donker PJ, Ivanovici F, Noach EL (1972) Analysis of the Urethral Pressure Profile by Means of Electromyography and the Administration of Drugs. Br J Urol 44: 180-193.
- Awad SA, Downie JW (1976) Relative Contributions of Smooth and Striated Muscles to the Canine Urethral Pressure Profile. Br J Urol 48: 347-354.
- Andersson KE, Sjogren C (1982) Aspects on the Physiology and Pharmacology of the Bladder and Urethra. Prog Neurobiol 19: 71-89.
- Gahlin K, Sparf B (1978) Differences in postsynaptic cx-adrenoceptor populations between isolated cat urethra and various other tissues. Acta Pharnac Toxicol 11: 48-55.
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein Measurement with the Folin Phenol Reagent. J Biol Chem 193: 265-275.
- Cheng Y, Prusoff WH (1973) Relationship Between the Inhibition Constant (Ki) and the Concentration of Inhibitor which causes 50 % Inhibition (150) of an Enzymatic Reaction. Biochem Pharmac 22: 3099-3108.
- Miach PJ, Dausse J-P, Meyer P (1978) Direct Biochemical Demonstration of Two Types of Adrenoceptor in Rat Brain. Nature 274: 492-494.
- Greenslade FC, Scorr CK, Chasin M, Madison SM, Tobia AJ (1979) Interaction of prazosin with alpha-adrenergic receptors - in vitro binding and in vivo Biochem Pharmac 28: 2409-2411.
- Hasegawa M, Townley RG (1982) Alpha and Beta-Adrenergic Receptors of Canine Lung Tissue Identification and Characterization of Alpha-Adrenergic Receptors by Two Different Ligands. Life Sci 30: 1035-1044.
- Ito H, Hoopes MT, Baum BJ, Roth GS (1982) K+ Release from Rat Parotid Cells: An al-Adrenergic Mediated Event. Biochem Pharm 31: 567-573.
- Hoffman BB, Lefkowitz RJ (1980) An Assay for Alpha-Adrenergic Receptor Subtypes Using [H]- Dihydroergocryptine. Biochem Pharm 29: 452-454.
- Weitzell R, Tanaka T, Starke K (1979) Pre- and Postsynaptic Effects of Yohimbine Stereoisomers on Noradrenergic Transmission in the Pulmonary Artery of the Rabbit. Naunyn Schmiedeberg's Arch Pharm 308: 127-136.
- Stiles GL, Hoffman BB, Hubbard M, Caron MG, Lefkowitz RJ (1983) Guanine Nucleotides and Alpha1 Adrenergic Receptors in the Heart. Biochem Pharm 33: 69-71.






