Journal of Oral Health and Dentistry (ISSN: 2638-499X)

Case Report
Dental Manifestations of Alagille Syndrome
5462
Views & Citations4462
Likes & Shares
The Alagille Syndrome (SAG), known as syndromic ductular paucity, is a multisystem hereditary disorder of autosomal dominant transmission with a prevalence of 1/70000 live births. SAG is caused by a mutation in the Jagged1 gene (JAG1), a ligand encoding a Notch receptor that plays an important role in determining the fate of the developing cell. SAG is characterized by the association of chronic cholestasis with cardiac, ocular, skeletal and facial features. Other signs described as minor may include renal, pancreatic, and oral manifestations. The objective of our article is to provide an update on this rare syndrome and dental management.
Keywords: Alagille syndrome, Cholestasis, Dental manifestations
INTRODUCTION
Alagille's syndrome (SAG) or syndromic ductal insufficiency [1] is an inherited multisystemic disorder of autosomal dominant transmission, characterized by chronic cholestasis related to insufficiency of the intrahepatic bile ducts, stenosis of the peripheral pulmonary arteries, vertebral anomalies, characteristic facies, posterior embryotoxon/lower segment abnormalities, retinopathy, pancreatic damage, and oral manifestations. We report a case of SAG in a young girl with structural dental malformations characterized by greenish-brown staining and hypo mineralization.
CASE REPORT
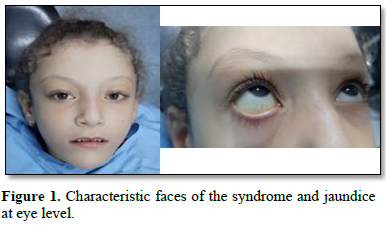
An 8-year-old girl was referred to our pediatric dentistry department, for management of anterior cross-bite. SAG had been diagnosed at birth with neonatal cholestasis associated with a common heterozygous JAG1 mutation. The girl presented all of the major criteria of AGS: cholestasis, bilateral embryotoxon and stenosis of the pulmonary artery branches associated to minor criteria: triangular face with slightly broad forehead and pointed chin (Figure 1).
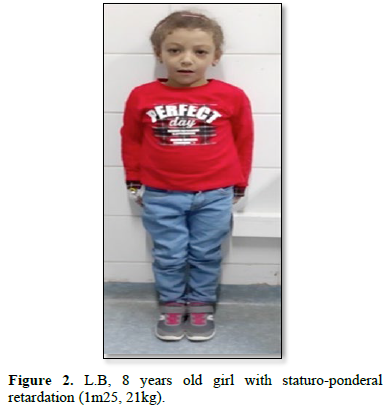
Biological tests showed significant cholestasis. Radiological studies showed a "butterfly-shaped" vertebra. The evolution was characterized by prolonged jaundice and significant growth retardation (Figure 2) [2]. Pruritus was controlled by symptomatic treatment: rifampicin, ursodeoxycholic acid, hydroxyzine and multivitamin supplementation.
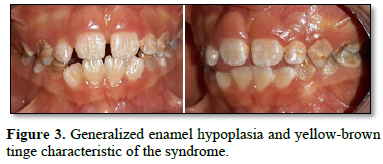
Dental examination showed a transitional dentition phase. Several oral abnormalities were observed with generalized
hypoplasia of the enamel characteristic of the syndrome with a yellow-brownish tinge, indicating the accumulation of bilirubin in the dental tissues during odontogenesis (Figure 3). Occlusion was also disturbed. Oral hygiene was insufficient and active carious lesions were observed on first molars and canine vestibular surfaces.
DISCUSSION
SAG is a rare genetic disorder defined as a disease associated with chronic cholestasis due to bile deficiency and the damage to several organs of varying severity [1]. Its prevalence is approximately 1/70,000 live births [1,2].
The disease can manifest itself in the newborn by prolonged jaundice due to conjugated hyperbilirubinemia and/or cardiac manifestations. Cardiac abnormalities include pulmonary atresia or stenosis, ventricular and/or atrial septal defect, tetralogy of Fallot and ductus arteriosus persistence [3,4]. Cholestasis is manifested by conjugated hyperbilirubinemia, hepatosplenomegaly, hypercholesterolemia, hypertriglyceridemia and coagulopathy. Pruritus and xanthomas may occur. Minor skeletal abnormalities include "butterfly wing" vertebrae (approximately 50% of cases) and shortening of the radii, ulna and phalanges. If present, facial dysmorphia usually appears in childhood and includes a prominent forehead, deep-set eyes in the sockets, palpebral slits pointing up and out, hypertelorism, a flat nose root and a pointed chin. Ophthalmic abnormalities include a posterior embryo toxin (75% of cases), Axenfeld's anomaly (see this term), retinitis pigmentosa, papillary or optic disc abnormalities. Growth retardation, fat malabsorption (rickets may occur) and sometimes developmental delay are present. Patients may have small dysplastic kidneys (common in SAG2) and hypothyroidism [5].
According to the severity of the hepatic disorders, the SAG can also reach the teeth, the salivary system, the glands, the periodontium and the mucous membranes. Dental manifestations are not a major criterion of the syndrome, but they inevitably occur as a complication of cholestasis and are related to hyperbilirubinemia. As a consequence of cholestasis during odontogenesis, opacification of the enamel, hypo mineralization and hypoplasia of the dental enamel may occur. In children with serum bilirubin levels above 30 mg/dl, bilirubin accumulates in dental tissue, causing variable greenish-brown dyschromia of the teeth. Oral manifestations may involve both temporary and permanent dentition [6]. Other authors have reported cases that present macrodontia of the maxillary incisors associated with taurodontism [7].
The treatment of the SAG is not specific, it includes a diet rich in carbohydrates and medium-chain triglycerides and a vitamin supplement. Pruritus is reduced by cholestyramine or rifampin. A liver transplant may be necessary in case of refractory disease. Cardiac or vascular procedures are considered for significant lesions [8].
Oral management should be done in collaboration with the pediatrician, who should inform us of the precautions to be taken before starting management, and prescribing an appropriate selection of medications and the use of antibiotics for prophylaxis or in the event of bleeding after hemostasis control by extraction. In the case of a liver transplant operation, all patients require regular dental check-ups due to permanent and continuous immunosuppression treatment and to avoid the risk of focal infection. Prolonged immunosuppression after transplantation may result in the removal of bone narrowness, which can lead to leukopenia, thrombocytopenia or anemia. It may predispose patients to excessive bleeding, opportunistic infections like mycosis, herpes superinfection, and development of leucoplakia. The use of cyclosporine can cause gingival hypertrophy, which can lead to gingivitis and lesions of the periodontal tissue. Regular dental care and prophylaxis, proper hygiene control and cyclosporine in consultation with the pediatrician, can prevent the appearance of these complications. In patients with AGS with less severe systemic manifestations, it is possible to achieve orthodontic treatment combined with cosmetic restorative procedures or surgery, but only with a monitoring of oral hygiene and control of caries is mandatory [9,10].
CONCLUSION
Dental disorders are not the major problems for people with AGS, but dentists, particularly pediatric dentists, may see patients with AGS in their daily practice. The most important points are careful observation, accurate diagnosis, and planned management of these patients by promoting multidisciplinary work. Preventive oral hygiene and consultations with medical specialists including pediatricians before any invasive procedure are mandatory. All this can improve the quality of life of patients with Alagille Syndrome.
CONSENT
Written informed consent was obtained from the patient for publication of this case report.
DISCLOSURE
This clinical case was written based on clinical observation without any funding.
CONFLICTS OF INTEREST
There are no conflicts of interest between the authors and between the authors and the patient.
- Jurkiewicz D, Popowska E, Gläser C, Hansmann I, Krajewska-Walasek M (2005) Twelve novel JAG1 gene mutations in Polish Alagille syndrome patients. Hum Mutat 25: 321.
- Alagille D, Estrada A, Hadchouel M, Gautier M, Odièvre M, et al. (1987) Syndromic paucity of interlobular bile ducts (Alagille syndrome or arteriohepatic dysplasia): Review of 80 cases. J Pediatr. 110(2): 195-200.
- Martinez AM, Cedillos CAM, Zermeno JN, Barroso VM, Carrión SV, et al. (2012) Dermatologic manifestations of Alagille syndrome. Bol Med Hosp Infant Mex 69(2): 132-136.
- Cheng K-W, Huang J-J, Wang C-H, Chen C-L, Jawan B (2004) Anesthetic management of a patient with Alagille’s syndrome undergoing living donor liver transplantation without blood transfusion. Chang Gung Med J 27(6): 449-453.
- Kamath BM, Schwarz KB, Hadzić N (2010) Alagille syndrome and liver transplantation. J Pediatr Gastroenterol Nutr 50(1): 11-15.
- Krantz ID, Piccoli DA, Spinner NB (1997) Alagille syndrome. J Med Genet 34(2): 152-157.
- Sokol RJ, Heubi JE, Balistreri WF (1983) Intrahepatic “cholestasis facies”: Is it specific for Alagille syndrome? J Pediatr 103(2): 205-208.
- Quiros-Tejeira RE, Ament ME, Heyman MB, Martin MG, Rosenthal P, et al. (1999) Variable morbidity in Alagille syndrome: A review of 43 cases. J Pediatr Gastroenterol Nutr 29(4): 431-437.
- Puja MR (2010) Oro-facial manifestation in Alagille syndrome. Int J Clin Dent 3(3): 219-224.
- Guadagni MG, Cocchi S, Tagariello T, Piana G (2005) Case report: Alagille syndrome. Minerva Stomatol 54(10): 593-600.
QUICK LINKS
- SUBMIT MANUSCRIPT
- RECOMMEND THE JOURNAL
-
SUBSCRIBE FOR ALERTS
RELATED JOURNALS
- Journal of Carcinogenesis and Mutagenesis Research (ISSN: 2643-0541)
- Journal of Infectious Diseases and Research (ISSN: 2688-6537)
- Archive of Obstetrics Gynecology and Reproductive Medicine (ISSN:2640-2297)
- Journal of Rheumatology Research (ISSN:2641-6999)
- Journal of Ageing and Restorative Medicine (ISSN:2637-7403)
- Journal of Psychiatry and Psychology Research (ISSN:2640-6136)
- Journal of Neurosurgery Imaging and Techniques (ISSN:2473-1943)



