BioMed Research Journal (ISSN:2578-8892)

Research Article
Maturity-Onset Diabetes of Youth Type 4: A Case Report of North of Iran
4623
Views & Citations3623
Likes & Shares
Genetically, diabetes can be categorized as polygenic or monogenic. Monogenic diabetes is a heterogeneous group of disorders including NDM, MODY and syndromic diabetes. HNF-4α (MODY 1), GCK (MODY 2) and HNF-1α (MODY 3) are the most common forms of MODY. MODY is known by this criterion; Dominant inheritance, NIDDM, and early onset. We report a rare type of MODY that is caused by IPF-1 (MODY 4) mutation.
Keywords: Monogenic diabetes, MODY, MODY 4, IPF-1
INTRODUCTION
Diabetes is generally classified into types 1 and 2, but in addition to these two types, there is a third category of diabetes secondary to non-genetic causes (such as medication, injury and also transplantation) or genetic. The latter type of diabetes is categorized as monogenic diabetes mellitus, which most neonatal diabetes mellitus (NDM) can be considered as, and maturity-onset diabetes of young (MODY) is the most common form of monogenic diabetes [1]. In monogenic diabetes, we have single mutations that can occur by dominant inheritance or due to sporadic mutations [2]. Typically, monogenic diabetes can manifest in three forms of NDM (which can itself be classified into three categories: Transient neonatal diabetes mellitus (TNDM), permanent neonatal diabetes mellitus (PNDM) and syndromic NDM), MODY, and diabetic syndromes. The different phenotypes of patients are related to abnormalities in the genes and their chromosomes [3]. MODY included about 5% of diabetes cases before the age of 45 years [4]. For the first time Tattersall et al. [5] reported a type of diabetes called MODY in 1974. The diabetes was in the mild form and inherited dominantly [6]. MODY genetic analysis was first performed in 1990, which identified five genes including glucokinase (GCK), hepatocyte nuclear factor (HNF4α), (HNF1α), insulin promoter factor (IPF) and HNF1β as the causes of MODY [7]. MODY is genetically defined as the autosomal dominant with high penetrance and non-insulin dependent (NIDDM) [6]. The classic findings of MODY include the absence of obesity and fat disorders such as dyslipidemia, which can cause insulin resistance. They often experience the disease in the 2nd to 4th decades, with a history of two successive generations of the disease [8]. To reduce the range of mutations involved in MODY and NDM, as well as to reduce the costs involved in treating patients, the Sanger sequencing method can be used to sequence common forms of mutation. Sequencing of genes is successful when we know the patients’ family as well as the patients’ symptoms [9-11], because of low frequency of this type of diabetes always they estimated lower than true incidences and so report of case can help for true diagnosis of then and also better management and consult for family.
CASE PRESENTATION
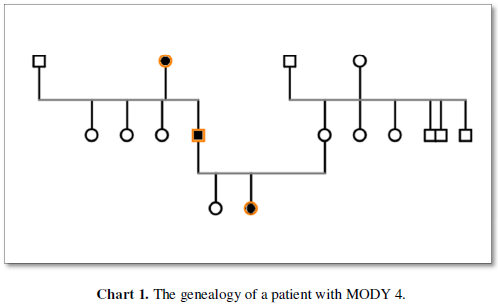
We report a patient that was diagnosed at age 14 as a case of MODY4. A 14-year-old girl with hyperglycemia was referred to our diabetes clinic. She was the first child of the family, full term, and was born by cesarean section. There was no history of abortion in her family. The patient’s birth weight was 4Kg and her height was 52cm. The duration of breastfeeding was up to 2 years old and complementary feeding was started at age 6 months. She also took vitamin A and D supplements up to 2 years. The patient had a history of hospitalization due to uncontrolled hyperglycemia that was not associated with ketoacidosis and seizure. The patient had no history of underlying disease, especially auto-immune disease. At the time of diagnosis, the patient weight was 57Kg and her BMI was 21.7Kg/m² she does not acanthuses, Nigerians. In the patient’s family history, her father had hyperlipidemia. On the other hand, there is a history of diabetes in the patient’s paternal grandmothers and in all paternal aunts. The patient had no history of weight loss, polyuria, or polydipsia. Her blood sugar profile included 2-hour post prandial blood glucose (2-hpp) or oral glucose tolerance test (OGTT) of 362mg/dl, fasting blood sugar (FBS) of 209mg/dl and hemoglobin A1C (HbA1c) of 11.5%. The patient’s lipid profile was total cholesterol of 148mg/dl, low-density lipoprotein (LDL) of 90mg/dl, high density lipoprotein (HDL) of 40mg/dl and triglyceride (TG) of 77mg/dl. Patient’s auto-antibodies profile including anti-tyrosine phosphatase ICA 512 (IA2), islet - cell antibodies (ICA) and anti-glutamic acid decarboxylase (GAD) were positive, with values of 13, 3.9 and 9.5, respectively. Also, at the time of diagnosis, her C-peptide was 2.3ng/ml. Urine analysis encompassed BUN of 14mg/dl, Creatinine of 0.9mg/dl and Urine/Specific Gravity (U/SG) of 1010. The patient was treated with Glibenclamide and Metformin because the first diagnosis was type 2 DM, then Glibenclamide was removed from the patient’s regimen and insulin Lantus was added. Due to the lack of precise control of blood sugar, Metformin, insulin Lantus and Novo Rapid were adjusted for the patient, currently being followed with HbA1c of 5.3%. Routine eye examinations were normal and there was no evidence of retinopathy and visual impairment. Renal function was also normal and albuminuria was not reported. It should be noted that the patient also has regular physical activity. Due to strong positive family history of DM, she was evaluated by genetic examine and the find diagnosis MODY 4 because of the result of PDX6, GATA 6 mutation (Chart 1).
DISCUSSION
Diabetes encompasses a wide range of metabolic disorders due to impaired insulin synthesis, glucose sensing, channelopathy, and impaired endoplasmic reticulum, impairing beta cell expression and function [2,12-14]. In general, diabetes is genetically polygenic, which includes type 1 and type 2 diabetes [12]. But monogenic form includes MODY, NDM (TNDM, PNDM) and syndromic diabetes [2,13,14]. Type 1 diabetes is an early form of autoimmune disease called insulin-dependent diabetes. On the other hand, we have non-insulin-dependent diabetes, which can be polygenic and monogenic, with type 2 diabetes being a type of non-insulin-dependent polygenic diabetes [12, 15]. Non-insulin-dependent monogenic diabetes including NDM, MODY, and rare diabetes-related syndromes [2, 13-15]. The most common mutations involved in NDM are in the KCNJ11 and ABCC8 genes, which are essential for the expression of the ATP-sensitive K-channel in beta cells [16-18]. The octameric structure of ATP-sensitive K-channel consists of two SUR1 and Kir6.2 subunits, which are encoded by the ABCC8 and KCNJ11 genes, respectively. Each subunit forms half of the central pole channel. The K-channels are blocked by the uptake of glucose metabolism and the increase of the ATP to ADP ratio in beta cells and then by stimulating beta cells and Ca input, insulin granules exocytosed. In fact, mutations involving the K-channel decrease the sensitivity of the channel and lead to open for a longer period of time, resulting in longer hyperglycemia [17]. MODY is one of the non-classic NIDDM, meaning there is a defect in insulin secretion and function. In fact, single gene mutations in beta cells cause the disease to progress slowly [19]. MODY is defined by a specific criterion that is based on the patient’s clinical presentation and symptom. Early onset: In fact, at last two family members under the age of 25 may be involved. This cut-off can be changed depending on the number of people in the family, diagnostic tests and anticipation. The anticipation phenomenon has caused that as more and more generations of the family get involved, the age of the disease decreases. With the development of diagnostic tests, the disease has been diagnosed at an early age [20,21]. On the other hand, involvement in a family member as well as in mild form of diabetes will be diagnosed at an older age, for example, in MODY 3, considering ages 10-60 years [22]. Non-insulin dependent: NIDDM is actually a form of diabetes that is defined by the specified amount of C-peptide or no need for insulin treatment within 5 years after diagnosis. This type of diabetes is suspected in cases of IDDM where a person’s hyperglycemic status can be maintained by diet and hypoglycemic agents over a honey moon period [20].
Inheritance: MODY inheritance is an autosomal type that can be dominant or pseudo-dominant. In the dominant inheritance, the single gene is involved. Whereas in the pseudo-dominant, polygenic disorders are involved. Both inheritances can be seen during pregnancy and vertical transmission. The early manifestations of NIDDM in family members are more indicative of a single gene. Autosomal dominant inheritance indicates a positive family history. It is said that most young people with diabetes who have no family history do not have a MODY diagnosis. But some patients miss out on lack of knowledge of their family history [21,23]. More than 10 genes are involved in MODY [24]. Mutations causing MODY are created on different chromosomes. Types 1 to 5 are due defects in chromosomes 20q, 7p, 12q, 13q and 17q, respectively [25]. MODY 1: HNF-4α acts as an HNF-1α regulator that plays a role in the function of hormone receptors in the steroid/thyroid branch [24,25]. This type of MODY is present at ages 7-15 years. Although insulin function is not a problem, the first and second phases of insulin secretion are gradually disrupted, with up to 30% of patients requiring insulin treatment over time due to insulin resistance [19,25]. MODY 2: The GCK gene can produce different types of diabetes depending on the type of mutation; MODY (heterozygous mutation) or NDM (homozygous mutation). This mutation creates the most common MODY variant [26,27]. This enzyme functions physiologically as a glucose sensor and plays a limiting enzyme role in the process of insulin secretion in response to glucose uptake in beta cells [28]. These patients have mild hyperglycemia that can progressively lead to insulin resistance, but the vascular complications are in expected because, this mutation is non-progressive [26,29-31]. The diagnosis of these patients is discussed before puberty with blood and urine (glycosuria) tests [30]. Depending on the mutation in the infant and the parents, the infant may have gain weight, loss weight or normal weight [32]. MODY 3: This is the most common variant of MODY in Asia, Europe and North America which is caused by HNF-1α mutation [29]. It is expressed in various organs such as the liver, kidney and pancreas [33,34]. Unlike GCK mutation, this is a progressive one that decreases pancreatic endocrine secretion in response to stimulation [24]. This MODY is expressed by abnormal glucose tolerance test (GTT) and insulin resistance in adulthood, which is not apparent before puberty [22]. Due to proximal tubule involvement, the nephropathy was in the form of glycosuria, aminoaciduria, and albuminuria [34]. MODY 4: Like HNF-α factor, the IPF-1 is involved in both the development and function of beta cells [35,36]. Mutation of this gene may produce different types of diabetes depending on the endocrine or exocrine involvement. If the disorder is purely endocrine disruption, MODY 4 is created and if it is both endocrine and exocrine, PNDM is created [37,38]. For the first time in 1997, MODY 4 was identified due to the heterozygous mutation of IPF-1 [36]. Of the endocrine regulators of the IPF-1, its role in the expression of the somatostatin, GCK, GLUT2 and pre-pro insulin genes has been noted [39]. The clinical signs of MODY 4 are similar to MODY 1 [40]. The most prominent characteristic of MODY 4 is hyperinsulinemia and obesity at an early age that cans misdiagnosis as type 2 DM [41]. In MODY diagnosis, clinical findings were used to confirm the disease and laboratory data were also used to rule out other types of diabetes. Clinical findings include age at onset of disease, non-insulin dependency, autosomal dominant inheritance, BMI, and laboratory data such as islet auto-antibody, OGTT and serum C-peptide levels [42,43]. The auto-antibodies used in the diagnosis of diabetes include antibody against islet antigen 2. Glutamic acid decarboxylase and islet cell [44]. Auto-antibodies are not a reliable criterion because patients have been found to have diabetes despite low auto-antibody titration and, On the other hand, auto-antibody titration decreases as the diseases progresses. C-peptide is not a sufficient criterion for diagnosis because in patients with early and chronic diabetes, a level of C-peptide was found discriminable [45]. In the HNF-4α mutation, all of the patients’ lipid profile decreased but LDL increased [46,47]. In patients with GCK mutation, an increase in HbA1c as well as decrease in lipid profile (HDL-cholesterol) is seen compared to the normal state [48]. Among patients with the HNF-1α and GCK mutations, patients with the HNF-1α mutation have significantly lower CRP [49]. Genetic analysis is performed on MODY patients when a young non-obese child with a positive family history] and abnormal tests such as FBS, OGTT and negative auto-antibody that does not experience any stress [50]. The evaluation of phenotypic variant of dominant gene has shown that common mutations cause less impairment in beta cell function and plasma glucose [51]. About 80% of MODY cases are caused by the three genes HNF-4α, HNF-1α and GCK [41]. Only these three genes are examined for MODY screening in pregnant women [33]. In the MODY patients, timely identification of patients with the aim of reducing vascular complications and insulin resistance is important [27]. One of the vascular complications in MODY patients is nephropathy. Among the MODY cases, mild hyperglycemia in MODY 2 rarely progresses to vascular complications [26,29]. MODY 1 and 4 due to poor control glycaemia are susceptible for leading to nephropathy [24,26,27,40]. MODY 3 patients have both micro and macro vascular complications [22,52]. On the other hand, renal involvement in these patients falsifies a syndrome similar to Fanconi [34]. MODY diagnosis can also be made on the basis of treatment, as young people with diabetes who are not obese respond to treatment with sulphonylureas [53]. In cases of k-channel mutations. Sulphonylurea is preferable to insulin because the patient experiences shorter periods of hypoglycemic. Also, a comparison of HbA1c induced by two drugs has shown that patients taking sulfonylurea have better glycemic control [16,54]. Treatment of patients with the GCK mutation does not decrease glucose levels and HbA1c because treatment of these patients decreases endogenous insulin secretion [55]. Treatment of patients with HNF-1α mutation is initiated with low dose sulphonyl urea [46]. If treatment with sulphonyl urea is not effective in patients with HNF-1α and HNF-4α mutations, insulin, dipeptidyl peptidase-4 (DPP-4) and glucagon-like peptide-1 (GLP-1) receptor agonist can be used [56]. Insulin and oral hypoglycemic agents (OHA) can be effective in patients with IPF-1 mutation [57,58]. As a result, depending on the genes mutations, patients develop different types of MODY that will have different approaches and treatments [59-62].
- American Diabetes Association (2015) Classification and diagnosis of diabetes. Diabetes Care 38(Suppl): S8-S16.
- Murphy R, Ellard S, Hattersley AT (2008) Clinical implications of a molecular genetic classification of monogenic beta-cell diabetes. Nat Clin Pract Endocrinol Metab 4: 200-213.
- Edghill EL, Hattersley AT (2008) Genetic disorders of the pancreatic beta cell and diabetes (permanent neonatal diabetes and maturity-onset diabetes of the young). In: Seino S, Bell GI (eds). Pancreatic Beta Cell in Health and Disease. Springer, Japan pp: 399-430.
- Thanabalasingham G, Pal A, Selwood MP, Dudley C, Fisher K, et al. (2012) Systematic assessment of etiology in adults with a clinical diagnosis of young-onset Type 2 diabetes is a successful strategy for identifying maturity-onset diabetes of the young. Diabetes Care 35: 1206-1212.
- Tattersall RB (1974) Mild familial diabetes with dominant inheritance. Q J Med 43: 339-357.
- Tattersall RB, Fajans SS (1975) A difference between the inheritance of classical juvenile-onset and maturity-onset type diabetes of young people. Diabetes 24: 44-53.
- Fajans SS, Bell GI (2011) MODY: History, genetics, pathophysiology, and clinical decision making. Diabetes Care 34: 1878-1884.
- Pearson ER, Pruhova S, Tack CJ, Johansen A, Castleden HAJ, et al. (2005) Molecular genetics and phenotypic characteristics of MODY caused by hepatocyte nuclear factor 4alpha mutations in a large European collection. Diabetologia 48(5): 878-885.
- Ku CS, Cooper DN, Polychronakos C, Naidoo N, Wu M, et al. (2012) Exome sequencing: Dual role as a discovery and diagnostic tool. Ann. Neurol 71: 5-14.
- Sule G, Campeau PM, Zhang VW, Nagamani SCS, Dawson BC, et al. (2013) Next-generation sequencing for disorders of low and high bone mineral density. Osteoporos Int 24: 2253-2259.
- Glöckle N, Kohl S, Mohr J, Scheurenbrand T, Sprecher A, et al. (2014) Panel-based next generation sequencing as a reliable and efficient technique to detect mutations in unselected patients with retinal dystrophies. Eur J Hum Genet 22: 99-104.
- McCarthy MI (2017) Genetics of T2DM in 2016: Biological and translational insights from T2DM genetics. Nat Rev Endocrinol 13(2): 71-72.
- Philipson LH, Murphy R, Ellard S, Hattersley AT, Stoy J, et al. (2015) Genetic diagnosis of endocrine disorders. In: Weiss, RE.; Refetoff, S., editors. Genetic Diagnosis of Endocrine Disorders. Elsevier London. pp: 472.
- Porter JR, Barrett TG (2005) Monogenic syndromes of abnormal glucose homeostasis: clinical review and relevance to the understanding of the pathology of insulin resistance and beta cell failure. J Med Genet 42: 893-902.
- Velho G, Froguel P (1997) The genetic determinants of NIDDM: strategies and recent results. Diabetes Metab 23: 7-17.
- Pearson ER, Flechtner I, Njolstad PR, Malecki MT, Flanagan SE, et al. (2006) Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. N Engl J Med 355(5): 467-477.
- Gloyn AL, Pearson ER, Antcliff JF, Proks P, Bruining GJ, et al. (2004) Activating mutations in the gene encoding theATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N Engl J Med 350(18): 1838-1849.
- Shepherd M, Pearson ER, Houghton J, Salt G, Ellard S, et al. (2003) No deterioration in glycemic control in HNF-1alpha maturity onset diabetes of the young following transfer from long-term insulin to sulphonylureas. Diabetes Care 26(11): 3191-3192.
- American Diabetes Association (2004) Diagnosis and classification of diabetes mellitus. Diabetes Care (2004) 27 (suppl): S5-S10.
- Tattersall RB, Mansell PJ (1991) Maturity onset diabetes of the young (MODY): One condition or many? Diabet Med 8: 402-410.
- Hattersley A (1997) Genetic factors in the etiology of non-insulin dependent diabetes. In: Leslie RDG (ed). Molecular pathogenesis of Diabetes Mellitus, 1st London: Karger. 22: 157-78.
- Isomaa B, Henricsson M, Lehto M, Forsblom C, Karanko S, et al. (1998) Chronic diabetic complications in patients with MODY3 diabetes. Diabetologia 41: 467-73.
- Frongel P, Zouali H, Vionnet N, Velho G, Vaxillaire M, et al. (1993) Familial hyperglycemia due to mutations in glucokinase: Definition of a subtype of diabetes mellitus. N Engl J Med 328: 697-702.
- Fajans SS, Brown MB (1993) Administration of sulfonylureas can increase glucose-induced insulin secretion for decades in patients with maturity onset diabetes of the young. Diabetes Care 16: 1254-1261.
- Polonski KS (1995) The β-cell in diabetes: From molecular genetics to clinical research. Diabetes 44: 705-717.
- Velho G, Froguel P (1998) Genetic, metabolic and clinical characteristics of maturity onset diabetes of the young. Eur J Endocrinol 138: 233-239.
- Njolstad PR, Oddmund S, Cuesta-Munoz A, Bjørkhaug L, Massa O, et al. (2001) Permanent neonatal diabetes mellitus due to glucokinase deficiency: an inborn error of the glucose/insulin signaling pathway. N Engl J Med 344: 1588-1592.
- Appleton M, Ellard S, Bulman M (1997) Clinical characteristics of the HNF-1α (MODY 3) and GCK mutations. Diabetologia 40: A161.
- Hattersley AT (1998) Maturity onset diabetes of the young: clinical heterogeneity explained by genetic heterogeneity. Diabet Med 15: 15-24.
- Clement K, Pueyo ME, Vaxillaire M, Rakotoambinina B, Thuillier F, et al. (1996) Assessment of insulin sensitivity in glucokinase-deficient subjects. Diabetologia 39: 82-90.
- Byrne MM, Sturis J, Clement K (1994) Insulin secretory abnormalities in subjects with hyperglycemia due to glucokinase mutations. J Clin Invest 93: 1120-1130.
- Colom C, Corcoy R (2010) Maturity onset diabetes of the young and pregnancy. Best Pract Res Clin Endocrinol Metab 24: 605-615.
- Yamagata K, Oda N, Kaisaki PJ, Menzel S, Furuta H, et al. (1996) Mutations in the hepatocyte nuclear factor-1 alpha gene in MODY 3. Nature 384: 455-458.
- Pontoglio M, Barra J, Hadchouel M, Doyen A, Kress C, et al. (1996) HNF 1α inactivation results in hepatic dysfunction phenylketonuria and renal Fanconi syndrome. Cell 84: 575-85.
- Stoffers DA, Zinkin NT, Stanojevic V, Clarke WL, Habener JF (1997) Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat Genet 15: 106-110.
- Stoffers DA, Ferrer J, Clarke WL, Habener JF (1997) Early-onset type-II diabetes mellitus (MODY4) linked to IPF1. Nat Genet 17: 138-139.
- Melloul D, Tsur A, Zangen D (2002) Pancreatic Duodenal Homeobox (PDX-1) in health and disease. J Pediatr Endocrinol Metab 15: 1461-1472.
- Thomas IH, Saini NK, Adhikari A, Lee JM, Kasa-Vubu JZ, et al. (2009) Neonatal diabetes mellitus with pancreatic agenesis in an infant with homozygous IPF-1 Pro63fsX60 mutation. Pediatr Diabetes 10: 492-496.
- Vaxillaire M, Frogue P (2008) Monogenic Diabetes in the Young, Pharmacogenetics and Relevance to Multifactorial Forms of Type 2 Diabetes. Endocrine Rev 29(3): 254-264.
- Bingham C, Ellard S, Allen L, Bulman M, Shepherd M, et al. (2000) Abnormal nephron development associated with a frameshift mutation in the transcription factor hepatocyte nuclear factor-1 beta. Kidney Int 57: 898-907.
- Fajans SS, Bell GI, Paz VP, Below JE, Cox NJ, et al. (2010) Obesity and hyperinsulinemia in a family with pancreatic agenesis and MODY caused by the IPF1 mutation Pro63fsX60. Transl Res 156: 7-14.
- Ellard S, Bellanne-Chantelot C, Hattersley AT (2008) Best practice guidelines for the molecular genetic diagnosis of maturity-onset diabetes of the young. Diabetologia 51: 546-553.
- Owen KR, Skupien J and Malecki MT (2009) The clinical application of non-genetic biomarkers for differential diagnosis of monogenic diabetes. Diabetes Res Clin Pract 86(Suppl 1): S15-S21.
- Howson JMM, Rosinger S, Smyth DJ, Boehm BO, ADBW-END Study Group, Todd JA (2011) Genetic analysis of adult-onset autoimmune diabetes. Diabetes 60: 2645-2653.
- Oram RA, Jones AG, Besser REJ, Knight BA, Shields BM, et al. (2014) The majority of patients with long-duration type 1 diabetes are insulin micro secretors and have functioning beta cells. Diabetologia 57(1): 187-191.
- Pearson ER, Pruhova S, Tack CJ, Johansen A, Castleden HAJ, et al. (2005) Molecular genetics and phenotypic characteristics of MODY caused by hepatocyte nuclear factor 4alpha mutations in a large European collection. Diabetologia 48: 878-885.
- Shih DQ, Dansky HM, Fleisher M, Assmann G, Fajans SS, et al. (2000) Genotype/phenotype relationships in HNF-4alpha/MODY1: Haploinsufficiency is associated with reduced apolipoprotein (AII), apolipoprotein (CIII), lipoprotein (a), and triglyceride levels. Diabetes 49: 832-837.
- Spegel P, Ekholm E, Tuomi T, Groop L, Mulder H, et al. (2013) Metabolite profiling reveals normal metabolic control in carriers of mutations in the glucokinase gene (MODY2). Diabetes 62(2): 653-661.
- Owen KR, Thanabalasingham G, James TJ, Karpe F, Farmer AJ, et al. (2010) Assessment of high-sensitivity C-reactive protein levels as diagnostic discriminator of maturity-onset diabetes of the young due to HNF1A mutations. Diabetes Care 33(9): 1919-1924.
- Owen K, Hattersley AT (2001) Maturity-onset diabetes of the young: from clinical description to molecular genetic characterization. Best Pract Res Clin Endocrinol Metab 15: 309-323.
- Consortium ST2 D, Estrada K, Aukrust I, Bjørkhaug L, Burtt NP, et al. (2014) Association of a low-frequency variant in hnf1a with type 2 diabetes in a latino population. JAMA 311(22): 2305-2314.
- Velho G, Vaxillaire M, Boccio V, Charpentier G, Froguel P (1996) Diabetes complications in NIDDM kindreds linked to the MODY3 locus on chromosome 12q. Diabetes Care 19: 915-919.
- Colclough K, Bellanne-Chantelot C, Saint-Martin C, Flanagan SE, Ellard S (2013) Mutations in the genes encoding the transcription factors hepatocyte nuclear factor 1 alpha and 4 alpha in maturity-onset diabetes of the young and hyperinsulinemic hypoglycemia. Hum Mutat 34: 669-685.
- Begum-Hasan J, Polychronakos C, Brill H (2008) Familial permanent neonatal diabetes with KCNJ11 mutation and the response to glyburide therapy – a three-year follow-up. J Pediatr Endocrinol Metab 21: 895-903.
- Stride A, Shields B, Gill-Carey O, Chakera AJ, Colclough K, et al. (2014) Cross-sectional and longitudinal studies suggest pharmacological treatment used in patients with glucokinase mutations does not alter glycaemia. Diabetologia 57: 54-56.
- Pearson ER, Badman MK, Lockwood CR (2004) Contrasting diabetes phenotypes associated with hepatocyte nuclear factor-1alpha and -1beta mutations. Diabetes Care 27: 1102-1107.
- Ryffel GU (2001) Mutations in the human genes encoding the transcription factors of the hepatocyte nuclear factor (HNF) 1 and HNF 4 families: functional and pathological consequences. J Molecular Endocrinol 27(1): 11-29.
- Iwasaki N, Okabe I, Momoi MY (2001) Splice site mutation in the hepatocyte nuclear factor-1 beta gene, IVS2nt+1G>A, associated with maturity onset diabetes of the young, renal dysplasia and bicormuate uterus. Diabetologia 44 (3): 387-388.
- Zamanfar D, Aarabi M, Amini M, Monajati M (2020) Prevalence of autoantibodies in type 1 diabetes mellitus pediatrics in Mazandaran, North of Iran. J Pediatric Endocrinol Metab 33(10): 1299-1305.
- Zamanfar D, Yazdani P, Aarabi M, Pournorooz H (2018) The prevalence of type 1 diabetes in children of Mazandaran province. Iran J Health Sci 6(2): 1-10
- Zamanfar D (2020) A case report of a patient with Maturity-Onset Diabetes of the Young type 9. Clin Excellence 9(3): 45-55.
- Zamanfar D, Aarabi M, Sadeghian I (2015) Type 1 diabetes mellitus associated with autoimmune thyroid disorders in Iranian children: A review. J Pediatr Rev 3(1).

QUICK LINKS
- SUBMIT MANUSCRIPT
- RECOMMEND THE JOURNAL
-
SUBSCRIBE FOR ALERTS
RELATED JOURNALS
- Advance Research on Endocrinology and Metabolism (ISSN: 2689-8209)
- Journal of Allergy Research (ISSN:2642-326X)
- Advance Research on Alzheimers and Parkinsons Disease
- Journal of Cancer Science and Treatment (ISSN:2641-7472)
- Journal of Carcinogenesis and Mutagenesis Research (ISSN: 2643-0541)
- Journal of Psychiatry and Psychology Research (ISSN:2640-6136)
- Journal of Otolaryngology and Neurotology Research(ISSN:2641-6956)


