International Journal of Surgery and Invasive Procedures (ISSN:2640-0820)

Research Article
Prenatal Diagnosis of Hemoglobinopathy by Chorionic Villi Sampling (CVS) in Odisha - The First Report
4092
Views & Citations3092
Likes & Shares
Objective: Hemoglobinopathies are the most common monogenic recessive disorders in India. Prenatal diagnosis and selective termination of an affected fetus is a feasible option to decrease the birth of affected babies. Lack of facilities in Odisha for prenatal diagnosis prompted us to establish the same in first trimester of pregnancy by chorionic villi sampling (CVS) analysis.
Methods: CVS was obtained between 10-13 weeks of gestation by transabdominal route with ultra sound guidance. Diagnosis was done by ARMS PCR to detect mutations for β-thalassemia and Sickle Cell Anemia.
Results: The present study includes45 cases of Hemoglobinopathy in first trimester of pregnancy from Odisha, a state situated in Eastern part of India. Fetal diagnosis showed:14 normal, 8HbS trait, 12b-thal trait, one HbE-trait, one db-thalassemia trait and 9 affected fetuses. All the couples agreed for termination of pregnancy having affected fetuses. The commonest mutation was IVS 1-5 (GàC) inb-thalassemia. Thirteen mothers of sickle syndrome include two cases of sickle thalassemia (S-bthal) and 11 cases of Sickle Cell Anemia (SS).
Conclusion: The prenatal diagnosis by CVS analysis utilizing DNA based technology has been successfully established for Hemoglobinopathy at Apollo Hospitals, Bhubaneswar. The type of mutation has also been identified. This facility will help in bringing down the birth of affected fetus (homozygous) of β-thalassemia and Sickle Cell Disease cases in Odisha.
Keywords: Haemoglobinopathies, Prenatal diagnosis, β-thalassmia, Sickle Cell Anemia INTRODUCTION
Hemoglobinopathy is the most common single gene disorder in India [1]. Odisha state is situated in Eastern part of India between West Bengal and Andhra Pradesh. There is paucity of reports about the exact incidence of b-thalassemia in Odisha. However, we see many cases of b-thalassemia major and homozygous sickle cell disease (personal observation). In order to control the birth of thalassemia major and homozygous SCD babies, prenatal screening by HPLC and subsequently prenatal diagnosis in the first trimester of pregnancy is very essential. In western part of Odisha, the high incidence of sickle cell Disease is reported [2,3]. Awareness about the prenatal diagnosis in SCD has been increased in the last decade. Genetic counselling for SCD has been shown to enjoy huge acceptance in parts of India like Gujrat, Maharashtra [4,5]. Consistent application of genetic counselling coupled with the awareness programme is essential for reduction of the prevalence of SCD in Odisha population [6]. There is no National programme of newborn screening for sickle cell disease in India till date. A homozygous child gets identified only when he or she becomes symptomatic. It is possible that many of the severely affected children die before the diagnosis is made. Awareness generation in interior regions in the last 5-10 yrs has probably led to increasing requests for prenatal diagnosis of the sickle syndromes [6].
In the present study, prenatal diagnostic facility for couples with hemoglobinopathy was established at Apollo Hospitals, Bhubaneswar, Odisha so that the couples at risk do not have to travel long to metros like Delhi or Mumbai. We offered prenatal diagnosis in the first trimester of pregnancy by CVS analysis using DNA based test.
MATERIALS & METHODS
Present study was approved by the Institutional Ethics Committee in the year 2014. The present data includes 45 cases of pregnant ladies. After proper genetic counselling explaining risks and complications associated with CVS procedure, informed and written consent in a prescribed Proforma was taken from the couples who gave consent for PND. After an informed consent, 3 ml EDTA blood of the couple and the affected child if any were collected for DNA analysis. Chorionic Villi Sampling (CVS) was performed by a trained Radiologist between 10-13 weeks of pregnancy with ultra sound guidance by transabdominal route under local anesthesia. The sample (20 to 30 mg Villi) was collected in sterile normal saline. The villus samples were cleaned under a dissecting microscope (Nikon NZ800) separating the maternal from fetal parts [7].
DNA extraction from chorionic Villi was done using spin column method (Qiagen). The quality of the sample was checked by spectrophotometer (Shimadzu, UV1800) method at A260/A280 ratio of 1.8 to 2. Then, ARMS-PCR was performed using Thermal cycler (ABI, Veriti) [8]. Amplification refractory system (ARMS) is a method for detecting any single base pair mutation or deletion. For each DNA sample ARMS PCR was put up in two tubes, one labelled as Normal (N) and other as mutant (M). In each tube we added a common forward primer, ARMS mixture and Taq polymerase. To check if the PCR system working properly and to avoid false negative result, a pair of internal control primers was added to each tube. Each of these primers was tested with known positive and negative control DNA samples under uniform and stringent PCR conditions. After PCR, the samples are run in 2% low melting agarose gel at 100 V for 1 h. The amplified products using normal and mutant primers for each mutation are run side by side for comparison. After electrophoresis, the gel observed under UV light in a gel documentation system. VNTR D1S80, ApoB, ACTBP2 were performed to check the maternal contamination [9].
RESULTS
During the period of four years from Aug, 2015-May, 2020, in 45 cases of prenatal diagnosis has been offered to the couples at risk. Some of the demographic data has been shown in Table 1.
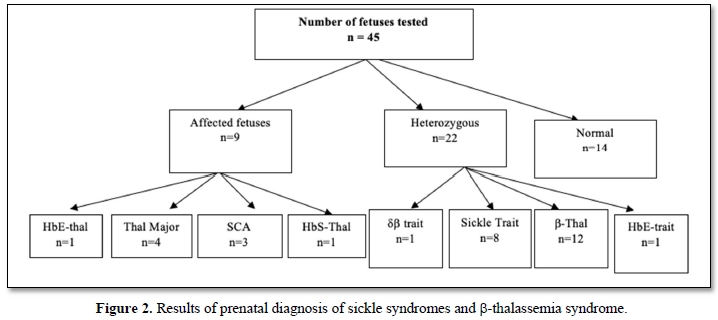
Majority of couples (80%) have one or more affected children. Only 9 couples (20 %) reported in the first trimester of pregnancy for prenatal diagnosis not having any child. The age group of the pregnant ladies varied from 25 to 40 years. Thirty (66.6%) ladies gave positive family history. In sickle cell cases majority belong to Western Odisha whereas in case of Thalassemia the couples hail from coastal districts of Odisha. The distribution of the same has been shown in Figure 1. The result of prenatal diagnosis has been shown in Figure 2.
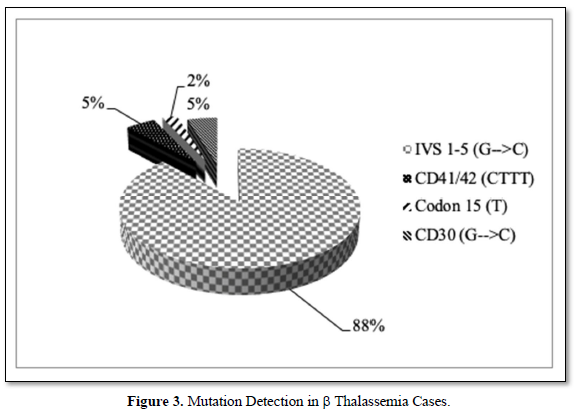
Nine fetuses were affected and twenty-two were heterozygous and ten were normal. Out of nine affected fetuses four were b-thalassemia, one HbE thalassemia and four were Sickle Cell Disease (SS-3& HbS-Thal-1). All the fetuses showing the presence of b-thalassemia gene had the most common Indian mutation IVS-1-5 (GàC) (Figure 3).
There was no fetal loss following CVS. All the couples with an unfavorable diagnosis opted for termination of pregnancy. A follow-up of all the 34 cases continuing pregnancies were tested from cord blood sampling during delivery by DNA method, i.e. ARMS-PCR for mutation to confirm the findings given earlier.
During this period one interesting case was observed who came for prenatal diagnosis of the second pregnancy (Figure 4).


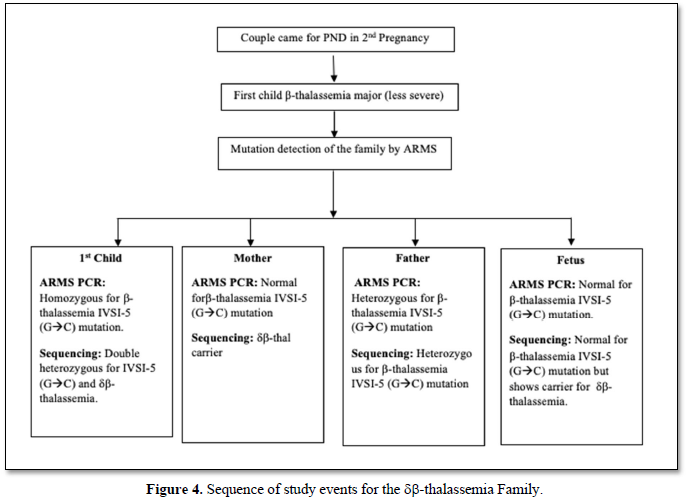
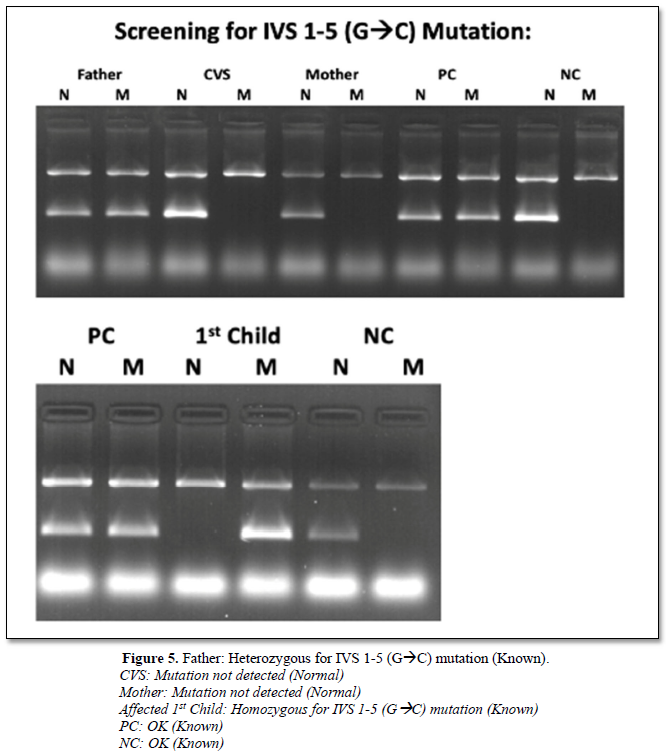
The first child was a thalassemia major whose severity was less. He was found to be homozygous for IVS 1-5 (GàC) (Figure 5).
The husband was found to have carrier status for IVS 1-5(GàC) and wife was normal for IVS 1-5 (GàC). So, the possibility of db thalassemia in the mother was considered. The peripheral blood picture of the mother was microcytic and hypochromic with low MCV and MCH. The CVS analysis showed normal pattern in agarose gel electrophoresis for 8 common mutations tested by ARMS technique. This is due to the presence of db-thal carrier status in the fetus, which was confirmed by sequencing b globin gene. The mother was allowed to continue the pregnancy.
DISCUSSION
The b-globin gene is a relatively small gene located in the short arm of chromosome 11. Although >180 causative mutations have been reported for b-thalassemia syndrome, the spectrum of mutations usually consists of a limited number of mutations and also some rare mutations in a particular community [10]. In India 8 common mutations including HbS and HbE have been found accounting to ~ 95% of all the mutations [11-15]. ARMS technique is an established reliable, accurate and rapid method in detecting these mutations and offering prenatal diagnosis within a period of 2 days [15].
In total the present study includes 45pregnant women where prenatal diagnosis has been offered. Out of these 14 fetuses are normal and 22 are heterozygous (Figure 2 and Table 1) and 9 affected fetuses. The mutation detected in b-thalassemia couples and fetuses are: IVS 1-5 (GàC) in 88% cases and three rare mutations accounting to 12%. Those rare mutations are CD41/42 (-CTTT), Codon 15 (-7) and CD30 (GàC) (Figure 3). The present study is in agreement with the published reports from other parts of our country that IVS 1-5 (GàC) is the most common mutation in b-thalassemia [11-14]. As expected, there are cases of sickleβ-thalassemia in Odisha as both b-thalassemia and Sickle Cell Disease (SCD) are prevalent in Odisha. In our small present study 2 cases of S-Thal and 13 cases of SS (homozygous) sickle cell are present. All the 25 β-thalassemia trait couples in our study, showed an IVSI-5 (GàC) in any one of the parents. Four cases of E-Thal when analyzed show IVS 1-5 (GàC) mutation in all of β-thalassemia trait parents along with CD26 (GàA). Identical mutations were present in both parents in 76.4 % of cases due to caste-based marriages in India.
It may be stressed here that the microcytic and hypochromic picture with normal iron status in hemogram is a good lead for investigating the presence of db thalassemia when b-thalassemia mutations are absent.
Prenatal diagnosis of b-thalassemia and other hemoglobinopathy from India has been reported earlier from different parts of India for control of birth of homozygous babies [3,15-17]. WHO (World Health Organization) recently published a prevalence map of SCD which showed that about 20-25 million people globally have HbSS; of this about 1-5 million live in Sub-Saharan Africa, 5-10 million live in the Indian subcontinent while about 3 million are distributed in other regions of the World [18]?
In USA with an equally significant burden of SCD genetic counselling and testing have been well established as early as 1970 and this has shown remarkable results in reducing the burden of homozygous sickle cell cases [19,20].
Antenatal screening programmes for hemoglobinopathies which is based on different ethnic origin have been similarly introduced in a number of European countries including Holland, Belgium and Germany with a good success rate [21].
The severity of sickle cell disease varies in India although majorities have Arab-Indian haplotype [22]. The reported birth rate of 1.1% for SCA (Sickle Cell Anemia) in central India with the highest incidence among populations in Mahar Community has been shown earlier [14]. Simultaneously the success story of prenatal diagnosis acceptance and termination of pregnancy in central India amongst the tribal community is very encouraging [3]. They found in their preliminary report that despite high prevalence of sickle cell gene the number of couples who underwent PND was less. This occurred mainly due to large number of women coming late for their first antenatal checkup, mostly during last trimester of pregnancy. We are sure that all these problems can be solved by genetic counselling before the test taking into account their belief, religious background and awareness among the general public.
Thus, prenatal diagnostic facility for cases of hemoglobinopathy is established in the state of Odisha successfully for the first time in a tertiary Health care Centre at Apollo Hospital, Bhubaneswar. It is interesting to note that b-thalassemias are prevalent in coastal districts and Sickle Cell Disease in Western and Southern Odisha. As opposed to other states of India, the prevalence of both sickle and β-thalassemia is found in Odisha and so also the cases of double heterozygous i.e. sickle thal and E-thal etc. (Figure 2). Some increased awareness amongst medical fraternity and general public will certainly bring down the load of hemoglobinopathy in the state of Odisha.
- Mohanty D, Colah RB, Gorakshakar AC, Patel RZ, Master DC, et al. (2013) Prevalence of β-thalassemia and other haemoglobinopathies in six cities in India: A multicentre study. J Commun Genet 1: 33-42.
- Colah R, Mukherjee M, Ghosh K (2014) Sickle cell disease in India. Curropin Hematol 21: 215-223.
- Singh PJ, Shrivastava AC, Shrikhande AV (2015) Prenatal diagnosis of sickle cell disease by the technique of PCR. Indian J Hematol Blood Transfus 21: 233-241.
- Italia Y, Krishnamurti L, Mehta V, Raicha B, Italia K, et al. (2015) Feasibility of a newborn screening and follow-up programme for sickle cell disease among South Gujrat (India) tribal populations. J Med Screen 22: 1-7.
- Jain DL, Sarathi V, Upadhye D, Gulhane R, Nadkarni AH, et al. (2012) Newborn screening shows a high incidence of sickle cell anemia in Central India. Haemoglobin 36: 316-322.
- Mohanty D, Das K (2011) Genetic counseling in tribals in India. Indian J Med Res 145: 561-571.
- Old JM (1986) Fetal DNA analysis, in genetic analysis of the human disease: A practical approach (Davies, K. E., ed.) IRL, Oxford, England, pp: 1-16.
- Old JM (2001) Disorders of hemoglobin-genetics, pathophysiology and clinical management. Cambridge: Cambridge University Press, pp: 941-948.
- Mohanty D, Colah R, Gorakshkar A (1994) Detection of b thalassemia mutation by amplification refractory mutation system (ARMS). Laboratory manual for screening diagnosis and molecular analysis of hemoglobinopathies and red cell enzymopathies. Bhalani Publishing House, Mumbai, pp: 98-101.
- Hardison RC, Chul DH, Glardine B, Riemer C, Patrinos G, et al. (2002) HbVar: A relational database of human hemoglobin variants and thalassemia mutations at the globin gene server. Hum Mutat 19: 225-233.
- Colah RB, Gorakshakar AC, Nadkarni AC, Nadkarni AH (2011) Invasive and non-invasive approaches for prenatal diagnosis of haemoglobinopathies: Experiences from India. Indian J Med Res 4: 552-560.
- Verma IC, Sexena R, Thomas E, Jain PK (1997) Regional distribution of beta-thalassemia mutations in India. Hum Genet 1: 109-113.
- Bandopadhy A, Bandyopathyay S, Chowdhury MD, Dasgupta UB (1999) Major b-globin gene mutations in Eastern India and their associated haplotypes. Hum Hered 49: 232-235.
- Agarwal S, Naveed M, Gupta UR, Kishore P, Agarwal SS, et al. (1994) Characterization of beta thalassemia mutation in 57 beta thalassemia families seen at Lucknow. Ind J Med Res 106-110.
- Saxena R, Jain PK, Thomas E, Verma IC (1998) Prenatal diagnosis of b-thalassemia: Experience in a developing country. Prenat Diagn 18: 1-7.
- Gorakshakar AC, Lulla CP, Nadkarni AH, Pawar AR, Desai SN, et al. (1997) Prenatal diagnosis of beta-thalassemia among Indians using denaturing gradient gel electrophoresis. Hemoglobin 5: 421-435.
- Colah R, GorakshaKar A, Phanasgaokar S, D’Souza E, Nadkarni A, et al. (2010) Epidemiology of b–thalassemia in Western India’ Mapping the frequencies and mutations in sub regions of Maharashtra and Gujrat. Br J Hematol 5: 739-747.
- Piel FB, Hay SI, Gupta S, Weatherall DJ, Williams TN, et al. (2013) Global burden of sickle cell anemia in children under five, 2010-2050: Modelling based on demographics, excess mortality and interventions. Plos Medicine 10: e1001484.
- Hassell KL (2010) Population estimates of sickle cell disease in the U S. Amer J Prev Med 4: S512-S521.
- Gallo AM, Wilkie D, Surez M, Labotka R, Molokie R, et al. (2010) Reproductive decisions in people with sickle cell disease or sickle cell trait. West J Nurs Res 32: 1073-1090.
- Modell B, Darlison M, Birgens H, Cario H, Faustino P, et al. (2007) Epidemiology of hemoglobin disorders in Europe. An overview. J Clin Lab Invest 67: 39-69.
- Jain D, Mohanty D (2018) Clinical manifestations of sickle cell disease in India: Misconceptions and reality. Curr Opin Hematol 3: 171-176.
-
 Table 1
Table 1






QUICK LINKS
- SUBMIT MANUSCRIPT
- RECOMMEND THE JOURNAL
-
SUBSCRIBE FOR ALERTS
RELATED JOURNALS
- Journal of Forensic Research and Criminal Investigation (ISSN: 2640-0846)
- Ophthalmology Clinics and Research (ISSN:2638-115X)
- Journal of Cell Signaling & Damage-Associated Molecular Patterns
- Dermatology Clinics and Research (ISSN:2380-5609)
- Journal of Cardiology and Diagnostics Research (ISSN:2639-4634)
- Oncology Clinics and Research (ISSN: 2643-055X)
- Journal of Clinical Trials and Research (ISSN:2637-7373)


