Proteomics and Bioinformatics (ISSN:2641-7561)

Research Article
Machine Learning Models for Drug Delivery in Subcellular Localization of HIV-1 subtypes and HIV-2
6756
Views & Citations5756
Likes & Shares
Proteins located in appropriate physiological contexts within a cell are a paramount importance in exerting their biological functions. Subcellular localization of proteins is essential as to maximize functional diversity and to find potential drug targets as many of the proteins are responsible for diseases. Conventional wet lab techniques to determine subcellular localization of proteins are costly, time consuming and laborious. So, in post genomic era the subcellular localization (SCL) prediction method needs some fast, accurate, achievable and promising methods. That’s why computational methods coupled with machine learning are used to predict and classify SCL of proteins of HIV-1 and its subtypes & HIV-2. Identification of SCL of proteins and classification with comparative analysis of different machine learning methods is achieved with great accuracy. This will bring new insights for drug delivery in diseases like HIV/AIDS.
Keywords: Subcellular localization, Genomic, Identification, Subtypes, Machine learning
INTRODUCTION
Acquired Immunodeficiency Syndrome (AIDS) is a life-threatening disease caused by Human Immunodeficiency Virus (HIV). The replication and recombination in HIV life cycle leads to small changes or mutation in sequence of HIV which shows many different forms of HIV, including within the body of a single person living with HIV [1]. Normally HIV is of two types, HIV type 1 and HIV type 2 are two distinct viruses. Worldwide the predominant virus is HIV-1.HIV-2 is mainly seen in a few West African countries. The spread in the rest of the world is negligible. Although HIV-2 generally progresses more slowly than HIV-1, some HIV drugs (NNRTIs like nevirapine and efavirenz) do not work against HIV-2.
On a structural level HIV-1 and HIV-2 have important genetic differences. The vpu gene found in HIV-1 is replaced by the vpx gene in HIV-2. In addition, the protease enzymes from the two viruses, which are aspartic acid proteases and have been found to be essential for maturation of the infectious particle, share about 50% sequence identity. Functionally their infection causing is different. HIV-1 enters the immune system by attaching onto the CD4+ receptor found on the surface of certain white blood cells. HIV-2 has a harder time for attachment. So, HIV-2 generally progresses much more slowly, with lower viral load and slower risk of becoming sick.
The strains of HIV-1 can be classified into four groups as discussed in figure1.The most important group M is the ‘major’ group and is responsible for the majority of the global HIV epidemic. The other three groups are N, O, P they are quite uncommon and only occur in Cameroon, Gabon and Equatorial Guinea. Within group M there are known to be at least 9 genetically distinct subtypes of HIV-1. These subtypes A, B, C, D, F, G, H, J, K. Additionally different subtypes can combine genetic material to form a hybrid virus, known as a ‘circulating recombinant forms (CRFs) of which few of them are identified [2] (Figure 1).
Viruses like HIV use host synthetic machinery to replicate hence called human immunodeficiency syndrome. They have evolved mechanisms to exploit the host nucleic acid replication and protein translation apparatus and have also developed strategies to evade humoral immune surveillance. Human immunodeficiency virus type 1 is a complex retrovirus encoding 15 distinct proteins. These are Gag and Env structural proteins MA (matrix), CA (capsid), NC (nucleocapsid), p6, SU (surface), and TM (transmembrane); the Pol enzymes PR (protease), RT (reverse transcriptase), and IN (integrase); the gene regulatory proteins Tat and Rev; and the accessory proteins Nef, Vif, Vpr, and Vpu. These proteins require targeting to appropriate subcellular compartments of the host cell to fulfill their roles. Experimentally viral proteins have shown localized in different cellular compartments including nucleus, mitochondria, plasma membrane etc. At present available drugs targets HIV enzymes and their entrance to human body.

Primary goal to ART includes:
- Reduce HIV-related morbidity and prolong survival
- Improve quality of life
- Restore and preserve immunologic function
- Maximally and durably suppress viral load
- Prevent vertical HIV transmission
Following Table gives the description of present drugs and their mode of action [5,6].
All these have many side effects like: nephrotoxicity, Fanconi’s syndrome, bone mineralization disorders (nrtis), rash, hepatotoxicity, neurocognitive impairment (efavirenz), teratogenicity (efavirenz) [nnrtis}, Potential for major drug interactions with numerous HIV and non-HIV drugs.

Hence there is a need to develop such potential sites for HIV drug delivery which are easy to predict and has no side effects afterwards. Protein subcellular localization prediction (or just protein localization prediction) is such a potential site which involves the prediction of where a protein resides in a cell, its subcellular localization. In general, prediction tools take as input information about a protein, such as a protein sequence of amino acids, and produce a predicted location within the cell as output, such as the nucleus, Endoplasmic reticulum, Golgi apparatus, extracellular space, or other organelles. The aim is to build tools that can accurately predict the outcome of protein targeting in cells. And it can aid the identification of drug targets. Experimentally determining the subcellular localization of a protein can be a laborious and time-consuming task. Immunolabeling or tagging (such as with a green fluorescent protein) to view localization using fluorescence microscope are often used. A high throughput alternative is to use prediction. Through the development of new approaches in computer science, coupled with an increased dataset of proteins of known localization, computational tools can now provide fast and accurate localization predictions for many organisms. This has resulted in subcellular localization prediction (SCL) becoming one of the challenges being successfully aided by bioinformatics, and machine learning.
Many prediction methods now exceed the accuracy of some high-throughput laboratory methods for the identification of protein subcellular localization [7]. Particularly, some predictors have been developed [8] that can be used to deal with proteins that may simultaneously exist, or move between, two or more different subcellular locations. Experimental validation is typically required to confirm the predicted localizations.
AUTHOR CONTRIBUTION
Looking into the importance of protein SCL for drug development. Here a study is being done for identification of SCL of HIV-1 M, N, O, P groups. To complete the study, HIV-1 M, N, O, Pgroups’ sequences were retrieved from Uniprot database and SCL of proteins are identified predicted by different protein subcellular localization prediction soft wares. And domain identification is done to find the relation between subcellular localization and target protein/domain.
ORGANIZATION OF WORK
The work can be organized as different methods for SCL prediction are described and tested for protein sequences of HIV subtypes, M, N, O, P and HIV-2 with identification and classification of SCL by Support Vector Machines. These are cross-validated and evaluated. Motif prediction and classification by machine learning technique is also done. Result and discussion completes the identification of SCL Proteins with their domains. Finally, conclusion and future scope of the present work.
METHOD
Computational methods for predicting protein subcellular localization can generally be divided into four categories: prediction methods based on (i) the overall protein amino acid composition, (ii) known targeting sequences, (iii) sequence homology and/or motifs, and (iv) a combination of several sources of information from the first three categories (hybrid methods). (i) The pioneering work in using the overall amino acid composition for prediction was done by Nakashima and Nishikawa, who presented a method for discriminating between intracellular and extracellular proteins [9]. Using the distance between the overall amino acid composition vectors, Cedano et al presented ProtLock for predicting five classes of subcellular localizations (extracellular, intracellular, integral membrane, anchor anchored membrane, and nuclear [9]. Reinhardt and Hubbard presented NNPSL, an approach using artificial neural networks (ANNs) for predicting four eukaryotic (cytoplasmic, extracellular, mitochondrial, and nuclear) and three prokaryotic (cytoplasmic, extracellular, and periplasmic) sub cellular localizations [10]. Several alternative algorithms have been applied to the data set presented by Reinhardt and Hubbard, including Kohonen’s self-organizing maps [11], Support Vector Machines [12-16] and Markov chain models [13]. Further developments in the area of using the overall amino acid composition have been made by reflecting sequence order effects. Chou et al presented an SVM-based methodfor predicting twelve different subcellular localizations, taking sequence order effects into account [14,15].
Software used for prediction of SCL in HIV are:
- EukMPloc: "Euk-mPLoc 2.0" is developed by hybridizing the gene ontology information, functional domain information, and sequential evolutionary information through three different modes of pseudo amino acid composition. It can be used to identify eukaryotic proteins among the following 22 locations: (1) acrosome, (2) cell wall, (3) centriole, (4) chloroplast, (5) cyanelle, (6) cytoplasm, (7) cytoskeleton, (8) endoplasmic reticulum, (9) endosome, (10) extracell, (11) Golgi apparatus, (12) hydrogenosome, (13) lysosome, (14) melanosome, (15) microsome (16) mitochondria, (17) nucleus, (18) peroxisome, (19) plasma membrane, (20) plastid, (21) spindle pole body, and (22) vacuole. Compared with the existing methods for predicting eukaryotic protein subcellular localization, this predictor is much more powerful and flexible, particularly in dealing with proteins with multiple locations and proteins without available accession numbers. For a newly-constructed stringent benchmark dataset which contains both single- and multiple-location proteins and in which none of proteins has pairwise sequence identity to any other in a same location, the overall jackknife success rate achieved by Euk-mPLoc 2.0 is more than 24% higher than those by any of the existing predictors. As a user-friendly web-server, Euk-mPLoc 2.0 is freely accessible at.
- Subloc: A server/client suite for protein subcellular location based on SOAP (Simple Object Access Protocol) technology. SubLoc facilitates automated information retrieval in adapting the SOAP technology to DBSubLoc database, a protein subcellular localization annotation database available through web browsers. This database contains more than 60 000 protein sequences from virus, bacteria, fungi, plant and animal.
- Virusploc: "Virus-PLoc" has been developed that is featured by fusing many basic classifiers with each engineered according to the K-nearest neighbor rule. The overall jackknife success rate obtained by Virus-PLoc in identifying the subcellular compartments of viral proteins was 80%. Virus-PLoc will be freely available as a web-server. Virus-PLoc has been used to provide large-scale predictions of all viral protein entries in Swiss-Prot database The results thus obtained have been deposited in a downloadable file prepared with Microsoft Excel and named "Tab_Virus-PLoc.xls." This file is available at the same website and will be updated twice a year to include the new entries of viral proteins and reflect the continuous development of Virus-PLoc.
- [A] To develop the methodology the dataset from Swissprot/Uniprot databank of Expasy server is obtained. The following four data sets were used.
Dataset 1: It consisted of all the subcellular locations of HIV-1 M group proteins predicted from Eukaryotic MPloc. The subcellular positions obtained are cell membrane, extracellular, cytoplasm, secreted proteins, nucleus and mitochondria. All the entries marked as fragments were not included in the dataset. The total instances were 280. The 280 were positive belonging to group and 280 were negative instances belonging to other enzymatic group. Table 1 shows individual position of subcellular location of HIV proteins. The predicted sites with their number of proteins are as- cell membrane=103, extracellular=6, cytoskeleton=7, cytoplasm=27, secreted proteins=22, Nucleus=94, Mitochondria=21.
Dataset 2: It consisted of all the subcellular locations of HIV-1N proteins predicted from Virus Ploc. The subcellular positions obtained are plasma membrane, extracellular, cytoplasm, nucleus and mitochondria. All the entries marked as fragments were not included in the dataset. The total instances were 200. The 200 were positive belonging to Virus Ploc and 200 were negative instances belonging to other enzymatic group. The predicted sites with number of proteins are as: Plasma membrane-108, Nucleus-74, Mitochondria-10, Cytoplasm-7, and Extracellular-1.
Dataset 3: It consisted of all the subcellular locations of HIV-1O proteins predicted from Sub loc software. The subcellular positions obtained are extracellular, cytoplasm, nucleus and mitochondria. All the entries marked as fragments were not included in the dataset. The total instances were 167. The 167 were positive belonging to Sub loc and 167 were negative instances belonging to other enzymatic group. The predicted sites with number of proteins are as: Extracellular-80, Cytoplasm-39, Nucleus-30, and Mitochondria-18.
Dataset 4: It consisted of all the subcellular locations of HIV-1P proteins predicted from Eukaryotic MPloc, Virus ploc and Sub loc software. The total instances were 100. The 100 were positive belonging to all the mentioned softwares and 100 were negative instances belonging to other enzymatic group. The predicted sites with number of proteins are as: Plasma membrane-40, Cytoplasm-28, Nucleus-20, and Mitochondria-12.
SUPPORT VECTOR MACHINE (BINARY CLASSIFICATION)
SVM is a supervised machine learning method which is based on the statistical learning theory [18-20]. When used as a binary classifier, an SVM will construct a hyperplane, which acts as the decision surface between the two classes. This is achieved by maximizing the margin of separation between the hyperplane and those points nearest to it. The SVMs were implemented using freely downloadable software, libSVM [18]. In this software there is a facility to define parameters and choose among various inbuilt kernals. They can be radial basis function (RBF) or a polynomial kernel (of given degree), linear, sigmoid.
SVM software; LIBSVM
Simulations were performed using LIBSVM version 2.89 (a freely available software package) [19]. For our study RBF Kernel was found to be the best. The SVM training was carried out by the optimization of the value of the regularization parameter and the value of RBF kernel parameter.
Amino Acid Composition
Previously, this parameter has been used for predicting the subcellular localization of proteins [20]. The amino acid composition is the fraction of each amino acid type within a protein.
The fractions of all 20 natural amino acids were calculated by using Equation 1,

Polycomp
The input vector of 450 was generated directly in the format of SVM by software Polycomp developed under Department of Bioinformatics, MANIT, Bhopal, India [21]. This software generates data which can be directly fed into the classifier hence saving valuable time and energy needed for formatting the hybrid.
Evaluation of Performance
The performance of our classifier was judged by 5-fold cross validation. The LibSVM provides a parameter selection tool using the RBF kernel: cross validation via grid search. A grid search was performed on C and Gamma using an inbuilt module of libSVM tools on Dataset 1, Dataset 2, Dataset 3 and Dataset 4 as shown in Figure 1. Here pairs of C and Gamma are tried and the one with the best cross validation accuracy.
[B]Known targeting sequences: Suppose HIV-1M group subtype C is targeted for subcellular localization then again sequences of HIV-1 Mgroup subtype C is identified is found to be 100 and subcellular localization of proteins are predicted by same software’s. The predicted sites with number of proteins are as: Plasma membrane-35, Cytoplasm-25, Nucleus-20, and Mitochondria-12, Secreted proteins-8. Same methodology of SVM and amino acid composition is applied and results are obtained as:
[C] Based on sequences homology or motif prediction of HIV-1 M, N, O, P groups following observations are obtained:
RESULT & DISCUSSION
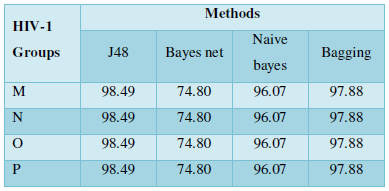
Protein Localization Prediction is important to predict the possible location or destination of a particular protein. It should be noted that this is still a prediction. An actual wet-lab procedure is preferred to do actual analysis of the protein's location. Since the prediction servers present possible location of the protein, then it also gives us insights of the possible function of the protein sequence. Since Eukaryotic MPloc, Subloc and Virus ploc are some of the software which predicts better results as shown in Tables 1-5.
Dataset 1: This includes HIV-1M group 280 sequences as identified by Uniprot protein database and their subcellular localization (SCL) of proteins are identified by EukMPloc,Subloc and Virusploc.
Dataset 2: This includes HIV-1N group 200 sequences as identified by Uniprot protein database and their subcellular localization (SCL) of proteins are identified by EukMPloc, Subloc and Virusploc.
Dataset 3: This includes HIV-1O group 167 sequences as identified by Uniprot protein database and their subcellular localization (SCL) of proteins are identified by EukMPloc, Subloc and Virusploc.
Dataset 4: This includes HIV-1 P group 100 sequences as identified by Uniprot protein database and their subcellular localization (SCL) of proteins are identified by EukMPloc,Subloc and Virusploc.
Dataset 5: This includes HIV-2 group 300 sequences as identified by Uniprot protein database and their subcellular localization (SCL) of proteins are identified by EukMPloc,Subloc and Virusploc.
Dataset 6: This includes HIV-1M group Subtype C 100 sequences as identified by Uniprot protein database and their subcellular localization (SCL) of proteins are identified by EukMPloc,Subloc and Virusploc.


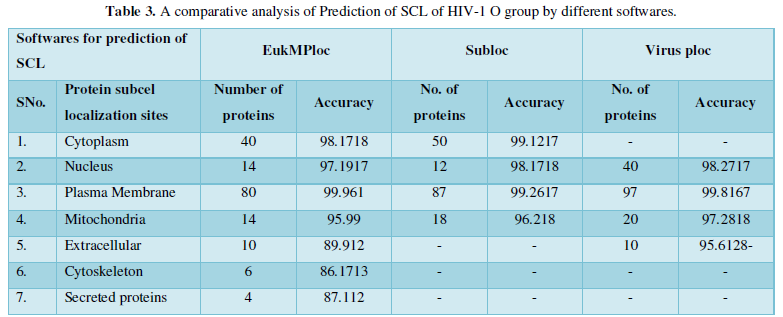


Subcellular localization prediction of all the datasets of HIV-1 M, N, O, P, HIV-2 and HIV-1Mgroup Subtype C shows cytoplasm, nucleus, plasma membrane and mitochondria would be the perfect SCL for HIV proteins. But EukMPloc is the only software which alone predicts extracellular space, cytoskeleton and secreted proteins as PSL sites. Hence it is said that all these sites play an important role in identifying potential drug target sites for HIV and its subtypes. This study will help biological scientists of pharma companies to manufacture HIV drugs or reformulate the drugs based on subcellular sites to avoid side effects. Subcellular sites of proteins as predicted by above methods and their role for drug target have been described as in Table 7.


A sophisticated prediction method should strive towards mimicking the biological process of protein sorting, an important step on the way to simulate small component of the systems biology of the cell. Machine learning plays an important role in the development and implementation of the complex underlying biological models, which can also be seen from the frequent application within this filed. As the accuracy of the methods continue to increase, it will become more interesting to take a closer look at the relatively small proportion of misclassified proteins. It is likely that these proteins are key players at the interfaces of the organelles and they may provide clues to alternative protein sorting routes. There is a need to constantly update methods and to extract new data sets for training the prediction models. To a general biologist, it is hard to assess the choice of algorithm and whether the accuracy estimation is done in a sound way. The pitfalls here are typically too homologous sequences within the training data and cross-validation schemas to estimate the overall performance of the method.
CONCLUSION
Understanding and predicting protein subcellular localization is a field of research whereas lot has happened by both experimentally and computationally. It is clear that several challenges lie ahead, especially when translating known facts from experimental biology into reasonable computational models and simultaneously to avoid simplifications. A further challenge is the prediction of proteins known to shuttle between compartments, which in principle can be ascribed to two subcellular localizations. There is no doubt that numerous new approaches, both in terms of algorithms and biological motivations, will be presented in this field in the near future. Prediction of subcellular localization is very likely to be one building block in systems biology approaches, aiming to understand the broader aspects of molecular biology. This new arena will open the development of SCL based molecular therapeutics for HIV-AIDS.
- Weiss RA (1993) “How does HIV cause AIDS?” Science 260(5112): 1273-1279.
- Douek DC, Roederer M, Koup RA (2009) “Emerging Concepts in the Immunopathogenesis of AIDS”. Ann Rev Med 60: 471-484.
- Powell MK, Benková K, Selinger P, Dogoši M, Luňáčková IK, et al. (2016) Opportunistic Infections in HIV-Infected Patients Differ Strongly in Frequencies and Spectra between Patients with Low CD4+ Cell Counts Examined Postmortem and Compensated Patients Examined Antemortem Irrespective of the HAART Era. PLOS One 11(9).
- Alison RJ, Cambiano V, Bruun T, Vernazza P, Collins S, et al. (2019) Risk of HIV transmission through condom less sex in serodifferent gay couples with the HIV-positive partner taking suppressive antiretroviral Bruun, therapy (PARTNER): final results of a multicentre, prospective, observational study. Lancet 393(10189): 2428-2438.
- Imamichi T (2004) Action of Anti-HIV Drugs and Resistance: Reverse Transcriptase Inhibitors and Protease Inhibitors. Curr Pharm Des 10(32): 4039-4053.
- Eric JA, Hazuda DJ (2012) HIV-1 Antiretroviral Drug Therapy. Cold Spring Harb Perspect Med 2(4): a007161.
- Rey S, Gardy JL, Brinkman FS (2005) Assessing the precision of high-throughput computational and laboratory approaches for the genome-wide identification of protein subcellular localization in bacteria. BMC Genomics 6: 162.
- Chou KC, Shen HB (2008) Cell-PLoc: A package of Web servers for predicting subcellular localization of proteins in various organisms. Nat Protoc 3(2): 153-162.
- Nakashima H, Nishikawa K (1994) Discrimination of intracellular and extracellular proteins using amino acid composition and residue-pair frequencies. J Mol Biol 238: 54-61.
- Cedano J, Aloy P, Pérez-Pons JA, Querol E (1997) Relation between amino acid composition and cellular location of proteins. J Mol Biol 266: 594-600.
- Reinhardt A, Hubbard T (1998) Using neural networks for prediction of the subcellular location of proteins. Nucleic Acid Res 26: 2230-2236.
- Cai YD, Chou KC (2000) Using neural networks for prediction of subcellular location of prokaryotic and eukaryotic proteins. Mol Cell Biol Res Commun 4: 172-173.
- Hua S, Sun Z (2001) Support vector machine approach for protein subcellular localization prediction. Bioinformatics 17: 721-728.
- Yuan Z (1999) Prediction of protein subcellular locations using Markov chain models. FEBS Lett 451: 23-26.
- Chou KC (2001) Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins 43: 246-255.
- Cai YD, Liu XJ, Xu XB, Chou KC (2002) Support vector machines for prediction of protein subcellular location by incorporating quasi-sequence-order effect. J Cell Biochem 84: 343-348.
- Chang CC, Lin CJ (2001) LIBSVM: A library for support vector machines.
- Vapnik V (1995) The nature of statistical learning theory, Springer.
- Wang M, Yang J, Liu GP, Xu ZJ, Chou KC (2004) Weighted support vector machines for predicting membrane protein types based on pseudo amino acid composition. Protein Eng Des 17: 509-516.
- Lavanya R, Mishra N, Pant B, Pant K, Pardasani KR (2010) ProCoS: Protein composition server Bioinformation 5(5): 227.
-
 Table 1
Table 1 -
Table 2
-
Table 3
-
Table 4
-
Table 5
-
Table 6
-
Table 7
-
Table 8
-
Table 9
-
Table 10











QUICK LINKS
- SUBMIT MANUSCRIPT
- RECOMMEND THE JOURNAL
-
SUBSCRIBE FOR ALERTS
RELATED JOURNALS
- Food and Nutrition-Current Research (ISSN:2638-1095)
- Journal of Biochemistry and Molecular Medicine (ISSN:2641-6948)
- Journal of Womens Health and Safety Research (ISSN:2577-1388)
- Journal of Microbiology and Microbial Infections (ISSN: 2689-7660)
- Journal of Veterinary and Marine Sciences (ISSN: 2689-7830)
- Journal of Genomic Medicine and Pharmacogenomics (ISSN:2474-4670)
- Journal of Agriculture and Forest Meteorology Research (ISSN:2642-0449)


